Questions about pre-computed SNIPER models #17
Comments
|
Hi, your script inputs are correct. I suspect the ValueError is because the autoencoder outputs a high-dimensional vector and the classifier expects a low-dimensional input. The all-B3 outputs you're seeing are likely because either:
|
|
Hi, Best, |
Sign up for free
to join this conversation on GitHub.
Already have an account?
Sign in to comment
Hi
Thanks to develop so wonderful tool, but I had some problems when I apply SNIPER models to my hic data.
When I have downloaded pre-computed SNIPER models, I found three kinds of files,
autoencoder/encoder/classifier. I have no idea to selectautoencoderorencoder, so I tried it all.When I used
autoencoderfile, I got error as followsand its output bed files looks very strange. Its all "B3" subcompartments through genome.
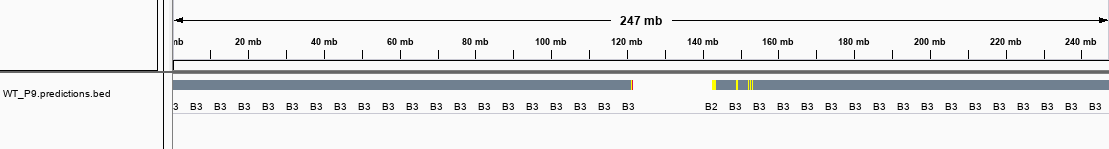
I have no idea to deal with it, Could you give me some sugesstions?
Thanks in advance!
Qianzhao
The text was updated successfully, but these errors were encountered: