The uPheno project aims to unify the annotation of phenotypes across species in a manner analogous to unification of gene function annotation by the Gene Ontology.
+uPheno 2.0 builds on earlier efforts with a strategy that directly leverages the work of the phenotype ontology development community and incorporates phenotypes from a much wider range of species.
+We have organised a collaborative community effort, including representatives of all major model organism databases, to document and align formal design patterns for representing phenotypes and further develop reference ontologies, such as PATO, which are used in these patterns.
+A common development infrastructure makes it easy to use these design patterns to generate both species-specific ontologies and a species-independent layer that subsumes them.
+The resulting community-curated ontology for the representation and integration of phenotypes across species serves two general purposes:
+- Providing a community-developed framework for ontology editors to bootstrap, maintain and extend their phenotype ontologies in a scalable and standardised manner.
+- Facilitating the retrieval and comparative analysis of species-specific phenotypes through a deep layer of species-independent phenotypes.
+
Currently, the development of uPheno is organized by a group that meets biweekly. See the meetings page for more info, including how to participate.
Matentzoglu N, Osumi-Sutherland D, Balhoff JP, Bello S, Bradford Y, Cardmody L, Grove C, Harris MA, Harris N, Köhler S, McMurry J, Mungall C, Munoz-Torres M, Pilgrim C, Robb S, Robinson PN, Segerdell E, Vasilevsky N, Haendel M. uPheno 2: Framework for standardised representation of phenotypes across species. 2019 Apr 8. http://dx.doi.org/10.7490/f1000research.1116540.1
EQ definitions are powerful tools for reconciling phenotypes across species and driving reasoning. However, they are not all that useful for many "normal" users of our ontologies.
+
We have developed a little workflow extension to take care of that.
We can now choose if we want to add the component to your edit file as well. To do that, follow the instructions on adding an import (i.e. adding the component to the edit file and catalog file). The IRI of the component is http://purl.obolibrary.org/obo/YOURONTOLOGY/components/eq-relations.owl. For example, for MP, the IRI is http://purl.obolibrary.org/obo/mp/components/eq-relations.owl.
+
Now we can generate the component:
+
sh run.sh make components/eq-relations.owl
+
+This command will be run automatically during a release (prepare_release).
The custom uPheno Makefile is an extension to your normal custom Makefile (for example, hp.Makefile, mp.Makefile, etc), located in the src/ontology directory of your ODK set up.
+
To install it:
+
(1) Open your normal custom Makefile and add a line in the very end:
Congiguring yamllint
+ You can ignore the error line too long yaml syntax errors for dos-dp yaml templates.
+ You can create a custom configuration file for yamllint in your home folder:
+
touch~/.config/yamllint/config
+
+ The content of the config file should look like this:
+
# Custom configuration file for yamllint
+# It extends the default conf by adjusting some options.
+
+extends:default
+
+rules:
+line-length:
+max:80# 80 chars should be enough, but don't fail if a line is longer
+# max: 140 # allow long lines
+level:warning
+allow-non-breakable-words:true
+allow-non-breakable-inline-mappings:true
+
+ The custom config should turn the error line too long errors to warnings.
Patternisation is the process of ensuring that all entity quality (EQ) descriptions from textual phenotype term definitions have a logical definition pattern. A pattern is a standard format for describing a phenotype that includes a quality and an entity. For example, "increased body size" is a pattern that includes the quality "increased" and the entity "body size." The goal of patternisation is to make the EQ descriptions more uniform and machine-readable, which facilitates downstream analysis.
+
1. Identify a group of related phenotypes from diverse organisms¶
+
The first step in the Phenotype Ontology Editors' Workflow is to identify a group of related phenotypes from diverse organisms. This can be done by considering proposals from phenotype editors or by using the pattern suggestion pipeline.
+The phenotype editors may propose a group of related phenotypes based on their domain knowledge, while the pattern suggestion pipeline uses semantic similarity and shared Phenotype And Trait Ontology (PATO) quality terms to identify patterns in phenotype terms from different organism-specific ontologies.
Once a group of related phenotypes is identified, the editors propose a phenotype pattern. To do this, they create a Github issue to request the phenotype pattern template in the uPheno repository.
+Alternatively, a new template can be proposed at a phenotype editors' meeting which can lead to the creation of a new term request as a Github issue.
+Ideally, the proposed phenotype pattern should include an appropriate PATO quality term for logical definition, use cases, term examples, and a textual definition pattern for the phenotype terms.
+
3. Discuss the new phenotype pattern draft at the regular uPheno phenotype editors meeting¶
+
The next step is to discuss the new phenotype pattern draft at the regular uPheno phenotype editors meeting. During the meeting, the editors' comments and suggestions for improvements are collected as comments on the DOS-DP yaml template in the corresponding Github pull request. Based on the feedback and discussions, a consensus on improvements should be achieved.
+The DOS-DP yaml template is named should start with a lower case letter, should be informative, and must include the PATO quality term.
+A Github pull request is created for the DOS-DP yaml template.
Once a consensus on the improvements for a particular template is achieved, they are incorporated into the DOS-DP yaml file. Typically, the improvements are applied to the template some time before a subsequent ontology editor's meeting. There should be enough time for off-line review of the proposed pattern to allow community feedback.
+The improved phenotype pattern candidate draft should get approval from the community at one of the regular ontology editors' call or in a Github comment.
+The ontology editors who approve the pattern provide their ORCIDs and they are credited as contributors in an appropriate field of the DOS-DP pattern template.
+
5. Add the community-approved phenotype pattern template to uPheno¶
+
Once the community-approved phenotype pattern template is created, it is added to the uPheno Github repository.
+The approved DOS-DP yaml phenotype pattern template should pass quality control (QC) steps.
+1. Validate yaml syntax: yamllint
+2. Validate DOS-DP
+Use DOSDP Validator.
+* To validate a template using the command line interface, execute:
+```sh
+yamllint
+dosdp validate -i
+
After successfully passing QC, the responsible editor merges the approved pull request, and the phenotype pattern becomes part of the uPheno phenotype pattern template collection.
The most convenient way to interact with S3 is the AWS Command Line Interface (CLI). You can find the installers and install instructions on that page (different depending on your Operation System):
+- For Mac
+- For Windows
Next, you need to ask someone at BBOP (such as Chris Mungall or Seth Carbon) to provide you with an account that gives you access to the BBOP s3 buckets. You will have to provide a username. You will receive:
+- User name
+- Access key ID-
+- Secret access key
+- Console link to sign into bucket
Now, you should be set up to write to your s3 bucket. Note that in order for your data to be accessible through https after your upload, you need to add --acl public read.
If you have previously pushed data to the same location, you wont be able to set it to "publicly readable" by simply rerunning the sync command. If you want to publish previously private data, follow the instructions here, e.g.:
Introduction to Continuous Integration Workflows with ODK¶
+
Historically, most repos have been using Travis CI for continuous integration testing and building, but due to
+runtime restrictions, we recently switched a lot of our repos to GitHub actions. You can set up your repo with CI by adding
+this to your configuration file (src/ontology/upheno-odk.yaml):
+
ci:
+ - github_actions
+
+
When updateing your repo, you will notice a new file being added: .github/workflows/qc.yml.
+
This file contains your CI logic, so if you need to change, or add anything, this is the place!
+
Alternatively, if your repo is in GitLab instead of GitHub, you can set up your repo with GitLab CI by adding
+this to your configuration file (src/ontology/upheno-odk.yaml):
+
ci:
+ - gitlab-ci
+
+
This will add a file called .gitlab-ci.yml in the root of your repo.
The editors workflow is one of the formal workflows to ensure that the ontology is developed correctly according to ontology engineering principles. There are a few different editors workflows:
+
+
Local editing workflow: Editing the ontology in your local environment by hand, using tools such as Protégé, ROBOT templates or DOSDP patterns.
+
Completely automated data pipeline (GitHub Actions)
+
DROID workflow
+
+
This document only covers the first editing workflow, but more will be added in the future
Ensure that there is a ticket on your issue tracker that describes the change you are about to make. While this seems optional, this is a very important part of the social contract of building an ontology - no change to the ontology should be performed without a good ticket, describing the motivation and nature of the intended change.
In your local environment (e.g. your laptop), make sure you are on the main (prev. master) branch and ensure that you have all the upstream changes, for example:
Create a new branch. Per convention, we try to use meaningful branch names such as:
+- issue23removeprocess (where issue 23 is the related issue on GitHub)
+- issue26addcontributor
+- release20210101 (for releases)
Using your editor of choice, perform the intended edit. For example:
+
Protégé
+
+
Open src/ontology/upheno-edit.owl in Protégé
+
Make the change
+
Save the file
+
+
TextEdit
+
+
Open src/ontology/upheno-edit.owl in TextEdit (or Sublime, Atom, Vim, Nano)
+
Make the change
+
Save the file
+
+
Consider the following when making the edit.
+
+
According to our development philosophy, the only places that should be manually edited are:
+
src/ontology/upheno-edit.owl
+
Any ROBOT templates you chose to use (the TSV files only)
+
Any DOSDP data tables you chose to use (the TSV files, and potentially the associated patterns)
+
components (anything in src/ontology/components), see here.
+
+
+
Imports should not be edited (any edits will be flushed out with the next update). However, refreshing imports is a potentially breaking change - and is discussed elsewhere.
+
Changes should usually be small. Adding or changing 1 term is great. Adding or changing 10 related terms is ok. Adding or changing 100 or more terms at once should be considered very carefully.
This step is very important. Rather than simply trusting your change had the intended effect, we should always use a git diff as a first pass for sanity checking.
+
In our experience, having a visual git client like GitHub Desktop or sourcetree is really helpful for this part. In case you prefer the command line:
+This will run the whole set of configured ODK tests on including your change. If you have a complex DOSDP pattern pipeline you may want to add PAT=false to skip the potentially lengthy process of rebuilding the patterns.
+
When you are happy with the changes, you commit your changes to your feature branch, push them upstream (to GitHub) and create a pull request. For example:
+
git add NAMEOFCHANGEDFILES
+git commit -m "Added biological process term #12"
+git push -u origin issue23removeprocess
+
+
Then you go to your project on GitHub, and create a new pull request from the branch, for example: https://github.com/INCATools/ontology-development-kit/pulls
+
There is a lot of great advise on how to write pull requests, but at the very least you should:
+- mention the tickets affected: see #23 to link to a related ticket, or fixes #23 if, by merging this pull request, the ticket is fixed. Tickets in the latter case will be closed automatically by GitHub when the pull request is merged.
+- summarise the changes in a few sentences. Consider the reviewer: what would they want to know right away.
+- If the diff is large, provide instructions on how to review the pull request best (sometimes, there are many changed files, but only one important change).
If you didn't run and local quality control checks (see 5a), you should have Continuous Integration (CI) set up, for example:
+- Travis
+- GitHub Actions
+
More on how to set this up here. Once the pull request is created, the CI will automatically trigger. If all is fine, it will show up green, otherwise red.
Once all the automatic tests have passed, it is important to put a second set of eyes on the pull request. Ontologies are inherently social - as in that they represent some kind of community consensus on how a domain is organised conceptually. This seems high brow talk, but it is very important that as an ontology editor, you have your work validated by the community you are trying to serve (e.g. your colleagues, other contributors etc.). In our experience, it is hard to get more than one review on a pull request - two is great. You can set up GitHub branch protection to actually require a review before a pull request can be merged! We recommend this.
+
This step seems daunting to some hopefully under-resourced ontologies, but we recommend to put this high up on your list of priorities - train a colleague, reach out!
When the QC is green and the reviews are in (approvals), it is time to merge the pull request. After the pull request is merged, remember to delete the branch as well (this option will show up as a big button right after you have merged the pull request). If you have not done so, close all the associated tickets fixed by the pull request.
It is sometimes difficult to keep track of changes made to an ontology. Some ontology teams opt to document changes in a changelog (simply a text file in your repository) so that when release day comes, you know everything you have changed. This is advisable at least for major changes (such as a new release system, a new pattern or template etc.).
We can define custom checks using SPARQL. SPARQL queries define bad modelling patterns (missing labels, misspelt URIs, and many more) in the ontology. If these queries return any results, then the build will fail. Custom checks are designed to be run as part of GitHub Actions Continuous Integration testing, but they can also run locally.
Add the SPARQL query in src/sparql. The name of the file should end with -violation.sparql. Please give a name that helps to understand which violation the query wants to check.
+
Add the name of the new file to odk configuration file src/ontology/uberon-odk.yaml:
+
Include the name of the file (without the -violation.sparql part) to the list inside the key custom_sparql_checks that is inside robot_report key.
+
+
If the robot_report or custom_sparql_checks keys are not available, please add this code block to the end of the file.
The documentation for UPHENO is managed in two places (relative to the repository root):
+
+
The docs directory contains all the files that pertain to the content of the documentation (more below)
+
the mkdocs.yaml file contains the documentation config, in particular its navigation bar and theme.
+
+
The documentation is hosted using GitHub pages, on a special branch of the repository (called gh-pages). It is important that this branch is never deleted - it contains all the files GitHub pages needs to render and deploy the site. It is also important to note that the gh-pages branch should never be edited manually. All changes to the docs happen inside the docs directory on the main branch.
All the documentation is contained in the docs directory, and is managed in Markdown. Markdown is a very simple and convenient way to produce text documents with formatting instructions, and is very easy to learn - it is also used, for example, in GitHub issues. This is a normal editing workflow:
+
+
Open the .md file you want to change in an editor of choice (a simple text editor is often best). IMPORTANT: Do not edit any files in the docs/odk-workflows/ directory. These files are managed by the ODK system and will be overwritten when the repository is upgraded! If you wish to change these files, make an issue on the ODK issue tracker.
+
Perform the edit and save the file
+
Commit the file to a branch, and create a pull request as usual.
+
If your development team likes your changes, merge the docs into master branch.
The documentation is not automatically updated from the Markdown, and needs to be deployed deliberately. To do this, perform the following steps:
+
+
In your terminal, navigate to the edit directory of your ontology, e.g.:
+
cd upheno/src/ontology
+
+
Now you are ready to build the docs as follows:
+
sh run.sh make update_docs
+
+ Mkdocs now sets off to build the site from the markdown pages. You will be asked to
+
Enter your username
+
Enter your password (see here for using GitHub access tokens instead)
+ IMPORTANT: Using password based authentication will be deprecated this year (2021). Make sure you read up on personal access tokens if that happens!
+
+
+
+
If everything was successful, you will see a message similar to this one:
+
INFO - Your documentation should shortly be available at: https://obophenotype.github.io/upheno/
+
+3. Just to double check, you can now navigate to your documentation pages (usually https://obophenotype.github.io/upheno/).
+ Just make sure you give GitHub 2-5 minutes to build the pages!
+
+
+
+
+
+
+
+
Ensure that all your pull requests are merged into your main (master) branch
+
Make sure that all changes to master are committed to GitHub (git status should say that there are no modified files)
+
Locally make sure you have the latest changes from master (git pull)
+
Checkout a new branch (e.g. git checkout -b release-2021-01-01)
+
You may or may not want to refresh your imports as part of your release strategy (see here)
+
Make sure you have the latest ODK installed by running docker pull obolibrary/odkfull
+
+
To actually run the release, you:
+
+
Open a command line terminal window and navigate to the src/ontology directory (cd upheno/src/ontology)
+
Run release pipeline:sh run.sh make prepare_release -B. Note that for some ontologies, this process can take up to 90 minutes - especially if there are large ontologies you depend on, like PRO or CHEBI.
+
If everything went well, you should see the following output on your machine: Release files are now in ../.. - now you should commit, push and make a release on your git hosting site such as GitHub or GitLab.
+
+
This will create all the specified release targets (OBO, OWL, JSON, and the variants, ont-full and ont-base) and copy them into your release directory (the top level of your repo).
(Optional) Rough check. This step is frequently skipped, but for the more paranoid among us (like the author of this doc), this is a 3 minute additional effort for some peace of mind. Open the main release (upheno.owl) in you favourite development environment (i.e. Protégé) and eyeball the hierarchy. We recommend two simple checks:
+
Does the very top level of the hierarchy look ok? This means that all new terms have been imported/updated correctly.
+
Does at least one change that you know should be in this release appear? For example, a new class. This means that the release was actually based on the recent edit file.
+
+
+
Commit your changes to the branch and make a pull request
+
In your GitHub pull request, review the following three files in detail (based on our experience):
+
upheno.obo - this reflects a useful subset of the whole ontology (everything that can be covered by OBO format). OBO format has that speaking for it: it is very easy to review!
+
upheno-base.owl - this reflects the asserted axioms in your ontology that you have actually edited.
+
Ideally also take a look at upheno-full.owl, which may reveal interesting new inferences you did not know about. Note that the diff of this file is sometimes quite large.
+
+
+
Like with every pull request, we recommend to always employ a second set of eyes when reviewing a PR!
Once your CI checks have passed, and your reviews are completed, you can now merge the branch into your main branch (don't forget to delete the branch afterwards - a big button will appear after the merge is finished).
Go to your releases page on GitHub by navigating to your repository, and then clicking on releases (usually on the right, for example: https://github.com/obophenotype/upheno/releases). Then click "Draft new release"
+
As the tag version you need to choose the date on which your ontologies were build. You can find this, for example, by looking at the upheno.obo file and check the data-version: property. The date needs to be prefixed with a v, so, for example v2020-02-06.
+
You can write whatever you want in the release title, but we typically write the date again. The description underneath should contain a concise list of changes or term additions.
When you are dealing with large ontologies, you need a lot of memory. When you see error messages relating to large ontologies such as CHEBI, PRO, NCBITAXON, or Uberon, you should think of memory first, see here.
Sometimes you will get cryptic error messages when using legacy tools using OBO format, such as the ontology release tool (OORT), which is also available as part of the ODK docker container. In these cases, you need to track down what axiom or annotation actually caused the breakdown. In our experience (in about 60% of the cases) the problem lies with duplicate annotations (def, comment) which are illegal in OBO. Here is an example recipe of how to deal with such a problem:
+
+
If you get a message like make: *** [cl.Makefile:84: oort] Error 255 you might have a OORT error.
+
To debug this, in your terminal enter sh run.sh make IMP=false PAT=false oort -B (assuming you are already in the ontology folder in your directory)
+
This should show you where the error is in the log (eg multiple different definitions)
+WARNING: THE FIX BELOW IS NOT IDEAL, YOU SHOULD ALWAYS TRY TO FIX UPSTREAM IF POSSIBLE
+
Open upheno-edit.owl in Protégé and find the offending term and delete all offending issue (e.g. delete ALL definition, if the problem was "multiple def tags not allowed") and save.
+*While this is not idea, as it will remove all definitions from that term, it will be added back again when the term is fixed in the ontology it was imported from and added back in.
+
Rerun sh run.sh make IMP=false PAT=false oort -B and if it all passes, commit your changes to a branch and make a pull request as usual.
Your ODK repositories configuration is managed in src/ontology/upheno-odk.yaml. Once you have made your changes, you can run the following to apply your changes to the repository:
+
sh run.sh make update_repo
+
+
There are a large number of options that can be set to configure your ODK, but we will only discuss a few of them here.
+
NOTE for Windows users:
+
You may get a cryptic failure such as Set Illegal Option - if the update script located in src/scripts/update_repo.sh
+was saved using Windows Line endings. These need to change to unix line endings. In Notepad++, for example, you can
+click on Edit->EOL Conversion->Unix LF to change this.
To add a new import, you first edit your odk config as described above, adding an id to the product list in the import_group section (for the sake of this example, we assume you already import RO, and your goal is to also import GO):
+
import_group:
+ products:
+ - id: ro
+ - id: go
+
+
Note: our ODK file should only have one import_group which can contain multiple imports (in the products section). Next, you run the update repo workflow to apply these changes. Note that by default, this module is going to be a SLME Bottom module, see here. To change that or customise your module, see section "Customise an import". To finalise the addition of your import, perform the following steps:
+
+
Add an import statement to your src/ontology/upheno-edit.owl file. We suggest to do this using a text editor, by simply copying an existing import declaration and renaming it to the new ontology import, for example as follows:
+
in your catalog, tools like robot or Protégé will recognize the statement
+in the catalog file to redirect the URL http://purl.obolibrary.org/obo/upheno/imports/go_import.owl
+to the local file imports/go_import.owl (which is in your src/ontology directory).
If you simply wish to refresh your import in light of new terms, see here. If you wish to change the type of your module see section "Customise an import".
A ROBOT filter module is, essentially, importing all external terms declared by your ontology (see here on how to declare external terms to be imported). Note that the filter module does
+not consider terms/annotations from namespaces other than the base-namespace of the ontology itself. For example, in the
+example of GO above, only annotations / axioms related to the GO base IRI (http://purl.obolibrary.org/obo/GO_) would be considered. This
+behaviour can be changed by adding additional base IRIs as follows:
If you wish to customise your import entirely, you can specify your own ROBOT command to do so. To do that, add the following to your repo config (src/ontology/upheno-odk.yaml):
Now feel free to change this goal to do whatever you wish it to do! It probably makes some sense (albeit not being a strict necessity), to leave most of the goal instead and replace only:
When running sh run.sh make update_repo, a new file src/ontology/components/mycomp.owl will
+be created which you can edit as you see fit. Typical ways to edit:
+
+
Using a ROBOT template to generate the component (see below)
+
Manually curating the component separately with Protégé or any other editor
+
Providing a components/mycomp.owl: make target in src/ontology/upheno.Makefile
+and provide a custom command to generate the component
+
WARNING: Note that the custom rule to generate the component MUST NOT depend on any other ODK-generated file such as seed files and the like (see issue).
+
+
+
Providing an additional attribute for the component in src/ontology/upheno-odk.yaml, source,
+to specify that this component should simply be downloaded from somewhere on the web.
Since ODK 1.3.2, it is possible to simply link a ROBOT template to a component without having to specify any of the import logic. In order to add a new component that is connected to one or more template files, follow these steps:
+
+
Open src/ontology/upheno-odk.yaml.
+
Make sure that use_templates: TRUE is set in the global project options. You should also make sure that use_context: TRUE is set in case you are using prefixes in your templates that are not known to robot, such as OMOP:, CPONT: and more. All non-standard prefixes you are using should be added to config/context.json.
+
Add another component to the products section.
+
To activate this component to be template-driven, simply say: use_template: TRUE. This will create an empty template for you in the templates directory, which will automatically be processed when recreating the component (e.g. run.bat make recreate-mycomp).
+
If you want to use more than one component, use the templates field to add as many template names as you wish. ODK will look for them in the src/templates directory.
+
Advanced: If you want to provide additional processing options, you can use the template_options field. This should be a string with option from robot template. One typical example for additional options you may want to provide is --add-prefixes config/context.json to ensure the prefix map of your context is provided to robot, see above.
Release file are the file that are considered part of the official ontology release and to be used by the community. A detailed description of the release artefacts can be found here.
Imports are subsets of external ontologies that contain terms and axioms you would like to re-use in your ontology. These are considered "external", like dependencies in software development, and are not included in your "base" product, which is the release artefact which contains only those axioms that you personally maintain.
Components, in contrast to imports, are considered full members of the ontology. This means that any axiom in a component is also included in the ontology base - which means it is considered native to the ontology. While this sounds complicated, consider this: conceptually, no component should be part of more than one ontology. If that seems to be the case, we are most likely talking about an import. Components are often not needed for ontologies, but there are some use cases:
+
+
There is an automated process that generates and re-generates a part of the ontology
+
A part of the ontology is managed in ROBOT templates
+
The expressivity of the component is higher than the format of the edit file. For example, people still choose to manage their ontology in OBO format (they should not) missing out on a lot of owl features. They may choose to manage logic that is beyond OBO in a specific OWL component.
One of the most frequent problems with running the ODK for the first time is failure because of lack of memory. This can look like a Java OutOfMemory exception,
+but more often than not it will appear as something like an Error 137. There are two places you need to consider to set your memory:
+
+
Your src/ontology/run.sh (or run.bat) file. You can set the memory in there by adding
+robot_java_args: '-Xmx8G' to your src/ontology/upheno-odk.yaml file, see for example here.
+
Set your docker memory. By default, it should be about 10-20% more than your robot_java_args variable. You can manage your memory settings
+by right-clicking on the docker whale in your system bar-->Preferences-->Resources-->Advanced, see picture below.
This page discusses how to update the contents of your imports, like adding or removing terms. If you are looking to customise imports, like changing the module type, see here.
Note: some ontologies now use a merged-import system to manage dynamic imports, for these please follow instructions in the section title "Using the Base Module approach".
+
Importing a new term is split into two sub-phases:
There are three ways to declare terms that are to be imported from an external ontology. Choose the appropriate one for your particular scenario (all three can be used in parallel if need be):
This workflow is to be avoided, but may be appropriate if the editor does not have access to the ODK docker container.
+This approach also applies to ontologies that use base module import approach.
+
+
Open your ontology (edit file) in Protégé (5.5+).
+
Select 'owl:Thing'
+
Add a new class as usual.
+
Paste the full iri in the 'Name:' field, for example, http://purl.obolibrary.org/obo/CHEBI_50906.
+
Click 'OK'
+
+
+
Now you can use this term for example to construct logical definitions. The next time the imports are refreshed (see how to refresh here), the metadata (labels, definitions, etc.) for this term are imported from the respective external source ontology and becomes visible in your ontology.
Every import has, by default a term file associated with it, which can be found in the imports directory. For example, if you have a GO import in src/ontology/go_import.owl, you will also have an associated term file src/ontology/go_terms.txt. You can add terms in there simply as a list:
You prefer to manage all your imported terms in a single file (rather than multiple files like in the "Using term files" workflow above).
+
You wish to augment your imported ontologies with additional information. This requires a cautionary discussion.
+
+
To enable this workflow, you add the following to your ODK config file (src/ontology/upheno-odk.yaml), and update the repository:
+
use_custom_import_module: TRUE
+
+
Now you can manage your imported terms directly in the custom external terms template, which is located at src/templates/external_import.owl. Note that this file is a ROBOT template, and can, in principle, be extended to include any axioms you like. Before extending the template, however, read the following carefully.
+
The main purpose of the custom import template is to enable the management off all terms to be imported in a centralised place. To enable that, you do not have to do anything other than maintaining the template. So if you, say currently import APOLLO_SV:00000480, and you wish to import APOLLO_SV:00000532, you simply add a row like this:
+
ID Entity Type
+ID TYPE
+APOLLO_SV:00000480 owl:Class
+APOLLO_SV:00000532 owl:Class
+
+
When the imports are refreshed see imports refresh workflow, the term(s) will simply be imported from the configured ontologies.
+
Now, if you wish to extend the Makefile (which is beyond these instructions) and add, say, synonyms to the imported terms, you can do that, but you need to (a) preserve the ID and ENTITY columns and (b) ensure that the ROBOT template is valid otherwise, see here.
+
WARNING. Note that doing this is a widespread antipattern (see related issue). You should not change the axioms of terms that do not belong into your ontology unless necessary - such changes should always be pushed into the ontology where they belong. However, since people are doing it, whether the OBO Foundry likes it or not, at least using the custom imports module as described here localises the changes to a single simple template and ensures that none of the annotations added this way are merged into the base file.
If you want to refresh the import yourself (this may be necessary to pass the travis tests), and you have the ODK installed, you can do the following (using go as an example):
+
First, you navigate in your terminal to the ontology directory (underneath src in your hpo root directory).
+
cd src/ontology
+
+
Then, you regenerate the import that will now include any new terms you have added. Note: You must have docker installed.
+
sh run.sh make PAT=false imports/go_import.owl -B
+
+
Since ODK 1.2.27, it is also possible to simply run the following, which is the same as the above:
+
sh run.sh make refresh-go
+
+
Note that in case you changed the defaults, you need to add IMP=true and/or MIR=true to the command below:
+
sh run.sh make IMP=true MIR=true PAT=false imports/go_import.owl -B
+
+
If you wish to skip refreshing the mirror, i.e. skip downloading the latest version of the source ontology for your import (e.g. go.owl for your go import) you can set MIR=false instead, which will do the exact same thing as the above, but is easier to remember:
+
sh run.sh make IMP=true MIR=false PAT=false imports/go_import.owl -B
+
Since ODK 1.2.31, we support an entirely new approach to generate modules: Using base files.
+The idea is to only import axioms from ontologies that actually belong to it.
+A base file is a subset of the ontology that only contains those axioms that nominally
+belong there. In other words, the base file does not contain any axioms that belong
+to another ontology. An example would be this:
The base file pipeline is a bit more complex than the normal pipelines, because
+of the logical interactions between the imported ontologies. This is solved by _first
+merging all mirrors into one huge file and then extracting one mega module from it.
+
Example: Let's say we are importing terms from Uberon, GO and RO in our ontologies.
+When we use the base pipelines, we
+
1) First obtain the base (usually by simply downloading it, but there is also an option now to create it with ROBOT)
+2) We merge all base files into one big pile
+3) Then we extract a single module imports/merged_import.owl
+
The first implementation of this pipeline is PATO, see https://github.com/pato-ontology/pato/blob/master/src/ontology/pato-odk.yaml.
+
To check if your ontology uses this method, check src/ontology/upheno-odk.yaml to see if use_base_merging: TRUE is declared under import_group
+
If your ontology uses Base Module approach, please use the following steps:
+
First, add the term to be imported to the term file associated with it (see above "Using term files" section if this is not clear to you)
+
Next, you navigate in your terminal to the ontology directory (underneath src in your hpo root directory).
+
cd src/ontology
+
+
Then refresh imports by running
+
sh run.sh make imports/merged_import.owl
+
+Note: if your mirrors are updated, you can run sh run.sh make no-mirror-refresh-merged
+
This requires quite a bit of memory on your local machine, so if you encounter an error, it might be a lack of memory on your computer. A solution would be to create a ticket in an issue tracker requesting for the term to be imported, and one of the local devs should pick this up and run the import for you.
+
Lastly, restart Protégé, and the term should be imported in ready to be used.
To add custom components to an ODK repo, please follow the following steps:
+
1) Locate your odk yaml file and open it with your favourite text editor (src/ontology/upheno-odk.yaml)
+2) Search if there is already a component section to the yaml file, if not add it accordingly, adding the name of your component:
5) Refresh your repo by running sh run.sh make update_repo - this should create a new file in src/ontology/components.
+6) In your custom makefile (src/ontology/upheno.Makefile) add a goal for your custom make file. In this example, the goal is a ROBOT template.
The Unified Phenotype Ontology (uPheno) meeting series¶
+
The uPheno editors call is held every second Thursday (bi-weekly) on Zoom, provided by members of the Monarch Initiative and co-organised by members of the Alliance and Genome Resources. If you wish to join the meeting, you can open an issue on https://github.com/obophenotype/upheno/issues with the request to be added, or send an email to phenotype-ontologies-editors@googlegroups.com.
+
The meeting coordinator (MC) is the person charged with organising the meeting. The current MC is Ray, @rays22.
The MC prepares the agenda in advance: everyone on the call is very busy and our time is precious.
+
Every agenda item has an associated ticket on GitHub, and a clear set of action items should be added in GitHub Tasklist syntax to the first comment on the issue.
+
If there are issues for any subtasks (e.g. PATO or Uberon edits), the list should be edited to link these.
+
Any items that do not have a subissue but do involve changes to patterns) should be edited to link to implementing PR.
+
It does not matter who wrote the first issue comment, the uPheno team can simply add a tasklist underneath the original comment and refine it over time.
+
Tag all issues which need discussion with "upheno call"
+
It must be clear from the task list what the uPheno team should be doing during the call (discuss, decide, review). For example, one item on the task list may read: "uPheno team to decide on appropriate label for template".
+
Conversely, no issue should be added to the agenda that does not have a clear set of action items associated with it that should be addressed during the call. These actions may include making and documenting modelling decisions.
+
Go through up to 10 issues on the uPheno issue tracker before each meeting to determine how to progress on them, and add action items. Only if they need to be discussed, add the "upheno call" label.
Every meeting should start with a quick (max 5 min, ideally 3 min) overview of all the goals and how they processed. The MC should mention all blockers and goals, even the ones we did not make any progress on, to keep the focus on the priorities:
+
uPheno releases
+
uPheno documentation
+
Pattern creation
+
Patternisation: The process of ensuring that phenotype ontologies are using uPheno conformant templates to define their phenotypes.
+
Harmonisation: The process of ensuring that phenotype patterns are applied consistently across ontologies.
+
For new pattern discussions:
+
Every new pattern proposal should come with a new GitHub issue, appropriately tagged.
+
The issue text should detail the use cases for the pattern well, and these use cases should also be documented in the "description" part of the DOSDP YAML file. Uses cases should include expected classifications and why we want them (and potentially classifications to avoid). e.g. axis-specific dimension traits should classify under more abstractly defined dimension traits which in term should classify under Morphology. Add some examples of contexts where grouping along these classifications is useful.
+
Agenda items may include discussion and decisions about more general modelling issues that affect more than one pattern, but these should also be documented as tickets as described above.
The uPheno organises an outreach call every four weeks to listen to external stakeholders describing their need for cross-species phenotype integration.
This includes a phenotype class for every anatomy class - the IRI is
+suffixed with "PHENOTYPE". Using these ontologies, Uberon and CL
+phenotypes make the groupings.
*http://www.human-phenotype-ontology.org/\
+* Köhler S, Doelken SC, Mungall CJ, Bauer S, Firth HV, Bailleul-Forestier I, Black GC, Brown DL, Brudno M, Campbell J, FitzPatrick DR, Eppig JT, Jackson AP, Freson K, Girdea M, Helbig I, Hurst JA, Jähn J, Jackson LG, Kelly AM, Ledbetter DH, Mansour S, Martin CL, Moss C, Mumford A, Ouwehand WH, Park SM, Riggs ER, Scott RH, Sisodiya S, Van Vooren S, Wapner RJ, Wilkie AO, Wright CF, Vulto-van Silfhout AT, de Leeuw N, de Vries BB, Washingthon NL, Smith CL, Westerfield M, Schofield P, Ruef BJ, Gkoutos GV, Haendel M, Smedley D, Lewis SE, Robinson PN. The Human Phenotype Ontology project: linking molecular biology and disease through phenotype data.Nucleic Acids Res.2014 Jan;42(Database issue):D966-74 [pubmed]
Currently the source is wbphenotype/wbphenotype-equivalence-axioms.obo,
+the OWL is generated from here. We are considering switching this
+around, so the OWL is edited, using Protege.
The ZP differs considerably from [HP], [MP] and others. ZFIN do not
+annotate with a pre-composed phenotype ontology - all annotations
+compose phenotypes on-the-fly using a combination of PATO, ZFA, GO and
+other ontologies.
+
We use these combinations to construct ZP on the fly, by naming each
+distinct combination, assigning it an ID, and placing it in the
+hierarchy.
"Characteristics" or "qualities" refer to an inherent or distinguishing characteristic or attribute of something or someone.
+It represents a feature that defines the nature of an object, organism, or entity and can be used to describe, compare, and categorize different things.
+Characteristics can be either qualitative (such as color, texture, or taste) or quantitative (such as height, weight, or age).
Characteristics such as the one above can be used to describe a variety of entities such as biological, environmental and social.
+We are specifically concerned with biological traits, which are characteristics that refer to an inherent characteristic of a biological entity, such as an organ (the heart), a process (cell division), a chemical entity (lysine) in the blood.
In biological contexts, the term "bearer" refers to the entity that possesses or carries a particular characteristic or quality.
+The bearer can be any biological entity, such as an organism, an organ, a cell, or even a molecular structure, that exhibits a specific trait or feature.
+Some examples:
+
+
Organism as a Bearer:
+
Example: A specific tree (such as an oak tree) is the bearer of the characteristic 'height'.
+
Explanation: The tree as an organism carries or has the property of height, making it the bearer of this characteristic.
+
+
+
Organ as a Bearer:
+
Example: The heart of a mammal can be the bearer of the characteristic 'heart size'.
+
Explanation: Here, the heart is the organ that possesses the 'heart size' charactertistic. The characteristic ('heart size') is a quality of the heart itself.
+
+
+
Cell as a Bearer:
+
Example: A red blood cell is the bearer of the characteristic 'cell diameter'.
+
Explanation: The diameter is a property of the individual cell. Thus, each red blood cell is the bearer of its diameter measurement.
+
+
+
Molecular Structure as a Bearer:
+
Example: A DNA molecule can be the bearer of the characteristic 'sequence length'.
+
Explanation: The length of the DNA sequence is a property of the DNA molecule itself, making the molecule the bearer of this characteristic.
+
+
+
Genetic Trait as a Bearer:
+
Example: A fruit fly (Drosophila melanogaster) can be the bearer of a genetic trait like eye color.
+
Explanation: The organism (fruit fly) carries the genetic information that determines eye color, making it the bearer of this specific trait.
+
+
+
+
In each example, the "bearer" is the entity that has, carries, or exhibits a particular biological characteristic. This concept is fundamental in biology and bioinformatics for linking specific traits, qualities, or features to the entities that possess them, thereby enabling a clearer understanding and categorization of biological diversity and functions.
A phenotypic change refers to some deviation from reference morphology, physiology, or behavior.
+This is the most widely used, and most complicated category of phenotype terms for data specialists to understand.
+
Conceptually, a phenotypic change comprises:
+
+
a biological attribute (which includes a biological bearer)
+
an "change" modifier
+
(optionally) a directional modifier (increased / decreased)
+
a comparator
+
+
Biological attributes such as blood lysine amount (OBA:2020005) have been discussed earlier in this document.
+The most widely used change modifier used in practice is abnormal (PATO:0000460).
+This modifier signifies that the phenotypic change term describes a deviation that is abnormal, such as "Hyperlysinemia" (HP:0002161), which describes and increased concentration of lysine in the blood.
+Other modifiers include normal (PATO:0000461), which describes a change within in the normal range (sometimes interpreted as "no change").
+A directional modifier like increased (PATO:0040043) or decreased (PATO:0040042). In practice, most of our "characteristic" terms have specialised directional variants such as decreased amount (PATO:0001997) which can be used to describe phenotypes.
Comparators are the most confusing aspects of phenotypic change.
+The first question someone has to ask when they see a concept describing is change like increased blood lysine levels is "compared to what?".
+Depending on biological context, the assumed comparators vary widely.
+For example, in clinical phenotyping, it is mostly assumed that
+a phenotypic feature corresponds to a deviation from the normal range, see HPO docs.
+However, it is just just as easily imaginable that HPO terms are used to describe change compared to a previous state of the same individual (increased tumor size compared to last time we checked).
+In research settings such as GWAS study annotations, HPO terms are used to annotate variants where a statistically significant change was observed compared to the general population.
+The same is true for many model phenotyping efforts such as MGI, where the situation is even further complicated that the comparator is not "the general population", but a control group. In summary, comparators can be:
+
+
The general population ("wild type" in research)
+
A non-representative sample of the general population (blood glucose values of all diabetes patients, a control group from the same mouse strain)
+
A previous state of a study subject (e.g. SNOMED).
+
+
And the compared charactertistics could be
+
+
Deviating from some notion of normality (abnormal)
+
A statistically significant change (including within normal)
+
+
No matter how much we want it - concepts describing phenotypic change will be used in many creative ways, and unfortunately, once the data hits your data analysis pipeline, you will likely not know for sure the nature of the comparator.
+Where you can, you should try to figure it out from the metata.
+
This sounds like bad news. However, keep one thing in mind:
+Phenotype associations (to anything, including genes) are rarely strictly causal.
+Even if a change is observed "compared to some non-representative control" there is likely to be some signal useful for downstream inference - somehow, the "gene has something to do with the phenotype".
In the clinical domain, many ontologies exist that define concepts that are very strongly related to our notion of "phenotype".
+In SNOMED, for example, "clinical findings" are defined as normal/abnormal observations, judgments, or assessments of patients (e.g. Abnormal urinalysis (finding)).
+For most analytic purposes, we think of SNOMEDs (and other medical terminologies) notion of clinical finding of something ortologous to our notion of "phenotype" (and their "observale entity" as a trait/biological attribute).
+However, if one gets into the weeds, many discrepencies in judgement can be observed, in particular when it comes to the separation from disease.
+
"Phenotype" is typically used in its "singular" form to describe the set of all observable characteristics of a subject.
+However, because we have over time gotten used to talking about "cardiovascular phenotype" and "increased blood glucose level", we have started using the plural form more, i.e. "phenotypes".
+We now tend to use the term "phenotypic profile" to describe the set of phenotypes that an organism exhibits at some point in time.
+
"Phenotypic feature" is a commonly used term that refers to the same idea, but mostly in the context of disease to describe an observable characteristic commonly associated with a disease.
+
"Phenotypic abnormality" is the formal term to describe a concept in the HPO, and is sometimes used to refer to the same idea in HPO-related papers.
+There is a bit of an assumption here, compared to the more general concepts described in this section, which is that the term should refer to a "deviation from the normal range", but, as described in the section of "implicit comparators", this assumption does not always hold in practice.
+
"Phenotypic change" is a recent invention by David Osumi-Sutherland in an attempt to subsume the ideas above, in particular to explicitly step back from the concept of "deviation from normal" to "statistically significant deviation" (which includes the normal range).
The Unified Phenotype Ontology (uPheno) is the reference ontology for biological abnormalities in the OBO world.
+There are a many species-specific ontologies in the OBO world, such as the Mammalian Phenotype Ontology (MP), the Human Phenotype Ontology (HPO) and the Drosophila Phenotype Ontology (DPO), see here.
Diseases are among the most important concepts in the phenotype data space. Phenotypes relate
+One big source of confusion in our community is the seperation of "phenotypic features" or changes from diseases.
+The HPO docs provide an explanation geared at clinicians to help them distinguish between the two.
+The quest on developing an operational definition is still ongoing, but for now, we recommend to go with the following basic assumptions:
+
+
There is a difference between disease and phenotype.
+
Phenotypes are features of diseases. Diseases can be associated with 1 or more phenotypic features. In the case of 1, we sometimes talk about "isolated X", for example "Isolated Growth Hormone Deficiency (IGHD)". IGHD is a condition where the pituitary gland produces insufficient growth hormone, leading to stunted growth.
+
Diseases, despite their grounding in biological reality, should be perceived mostly as social constructs that (adjusted from HPO docs):
+
Are used to capture a diagnosis (not just an observation - a judgement).
+
Are associated with a defined etiology (whether identified or as yet unknown, idiopathic). This is not about have some cause. This is about having a specific cause, even if it is unknown.
+
Have a defined time course (more or less well understood).
+
If treatments exist, there is a characteristic response to them.
+The key point is that the above are part of the disease definition.
In biological data curation, it’s essential to differentiate between traits (observable characteristics such as "blood glucose level") and measurements (a process to observe such characteristics, e.g. "blood glucose level assay", "BMI").
+Just from the term itself this is often difficult.
+"Blood glucose level" can refer both a measurement and a trait when taken out of context, but the ontologies they appear in should differenciate cleanly between the two.
+Here are some ways to distinguish them:
+- traits are
+ - observable characteritics of an organism
+ - can be qualitative ("red eye colour") or quantitative ("35 cm tail length")
+- measurements are
+ - activties performed by an agent (such as a researcher)
+ - involve the quantification or qualification of a specific trait
+ - correspond to measurement instruments / techniques (such as assays, BMIs)
+
In practice, it is true that a lot of data records a wild mix of the two.
+It is the job of (semantic) data modeling specialists to clearly distinguish the two when integrating annotate data from sources with divergent curation practices.
Characteristics (A) and bearers of characteristics (B) are the core constituents of traits/biological attributes (C). Phenotypes are comprised of trait terms (C) combined with a modifier (D). Species-specific phenotypes (F), including phenotypic abnormalities defined in the Human Phenotype Ontology (HPO) are feature of diseases (G). Measurements (H), such as assays, quantify or qualify (measure) traits (C).
Before we get started, let's remind ourselves of the basic structure of phenotype data.
+
+
Characteristics (A) and bearers of characteristics (B) are the core constituents of traits/biological attributes (C). Phenotypes are comprised of trait terms (C) combined with a modifier (D). Species-specific phenotypes (F), including phenotypic abnormalities defined in the Human Phenotype Ontology (HPO) are feature of diseases (G). Measurements (H), such as assays, quantify or qualify (measure) traits (C).
+
Integrating all kinds of phenotype data into the "uPheno framework" is a complex process which we will break down in the following.
To avoid multiple duplicate classes for heart, lung, skin etc we map all
+classes to [Uberon] where this is applicable. For more divergent species
+such as fly and C elegans we use the appropriate species-specific
+ontology.
+
Currently there are a small number of highly specific classes in FMA
+that are being used and have no corresponding class in Uberon
The current design patterns are such that the abnormal qualifier is only
+added when the quality class in the definition is neutral.
+
However, we still need to be able to infer
+
* Hyoplasia of right ventricle SubClassOf Abnormality of right ventricle
+
Because the latter class definition includes qualifier some abnormal,
+the SubClassOf axiom will not be entailed unless the qualifier is
+explicitly stated or inferred
Much has been written on the subject of representing absence. Before
+diving into the logical issues it is worth examining patterns in
+existing phenotype ontologies to understand what user expectations may
+typically be for absence.
From a strict logical perspective, this is inverted. "absent incisors"
+surely means "absence of all incisors", or put another way "the animal
+has no incisors". Yet it would be possible to have an animal with
+*absent* lower incisors and *present* upper incisors, yielding what
+seems a contradiction (because the subClass axiom would say this
+partial-incisor animal lacked all incisors).
+
If the ontology were in fact truly modeling "absence of *all* S" then
+it would lead to a curious ontology structure, with the typical tree
+structure of the anatomy ontology representing S inverted into a
+polyhierarchical fan in the absent-S ontology.
+
From this it can be cautiously inferred that the intent of the phenotype
+ontology curator and user is in fact to model "absence of *some* S"
+rather than "absence of *all* S". This is not necessarily a universal
+rule, and the intent may vary depending on whether we are talking about
+a serially repeated structure or one that typically occurs in isolation.
+The intent may also be to communicate that a *significant number* of S
+is missing.
Again, from a strict logical perspective this is false. If the spleen is
+absent then what does the "morphology" of the parent refer to?
+
However, this inference is clearly a desirable one from the point of
+view of the phenotype ontology editors and users, as it is common in
+ontologies for a variety of structures. For example:
+
And:
+
These patterns can be formally defended on developmental biology
+grounds. "absence" here is _not_ equivalent to logical absence. It
+refers specifically to developmental absence.
+
Furthermore, strict logical absence leads to undesirable inferences. It
+would be odd to include a nematode worm as having the phenotype "spleen
+absent", because worms have not evolved spleens. But the logical
+description of not having a spleen as part fets a worm.
+
Similarly, if the strict cardinality interpretation were intended, we
+would expect to see:
+
i.e. if you're missing your entire hindlegs, you're *necessarily*
+missing your femurs. But it must be emphatisized that this is *not*
+how phenotype ontologies are classified. This goes for a wide range of
+structures and other relationship types. In MP, "absent limb buds" are
+*not* classified under "absent limbs", even though it is impossible
+for a mammal to have limbs without having had limb buds.
The existing treatment of absence can be formally defended
+morphologically by conceiving of a morphological value space, with
+"large" at one end and "small" at the other. As we get continuously
+smaller, there may come an arbitrary point whereby we say "surely this
+is no longer a limb" (and of course, we are not talking about a pure
+geometrical size transformation here - as a limb reaches extreme edges
+of a size range various other morphological changes necessarily happen).
+But this cutoff is arguably arbitrary, and the resulting discontinuity
+causes problems. It is simpler to treat absence as being one end of a
+size scale.
This is barely touching the subject, and is intended to illustrate that
+things may be more subtle than naively treating words like "absent" as
+precisely equivalent to cardinality=0. An understanding of the medical,
+developmental and evolutionary contexts are absolutely required,
+together with an understanding of the entailments of different logical
+formulations.
+
Even though existing phenotype ontologies may not be conceived of
+formally, it is implicit than they do not model absence as being
+equivalent to cardinality=0 / not(has_part), because the structure of
+these ontologies would look radically different.
Pre-coordinated phenotype data is popular in the clinical domain, where a lot of observations are taken by a clinician and recorded as "phenotypic abnormalities" with the goal of eventual diagnosis.
+
Phenopackets such as the one below are an emerging standard to capture and sharing disease and phenotype information. Phenotypic features in particular are captured as so called "pre-coordinated phenotype terms" such as "Attenuation of retinal blood vessels" (HP:0007843). "Pre-coordinated" in this context means that the various aspects of the phenotype term, such as the bearer ("retinal blood vessels") and the characteristic ("Attenuation", or "thinning/narrowing"), and the modifier (in the case of HPO terms, simply abnormal), are combined ("coordinated") into a single term.
Apart from clinical diagnostics, pre-coordinated phenotype terms are used in many other contexts such as model organism research (e.g. IMPC) or the curation of Genome Wide Association Studies.
Post-coordinated phenotype curation simply means that the different constituents of phenotype (characteristic, bearer, modifier etc) are captured individually.
+This has certain advantages.
+For example, the phenotype space is enormous, as you can measure variations in many observable charactertics from chemical entities present in the blood, the microbiome to a host of morphological and developmental abnormalities. Instead of having individual (controlled vocabulary) terms for increased level of X, decreased level X, abnormal level of X, increased level of X in blood for thousands of chemical compounds synthesized by the human body, you just have "increased level", "blood" and all the chemical compounds.
+
There are at least three flavours of post-coordinated phenotype curation prevalent in the biomedical domain, four if you count quantified phenotypes:
The bearer-only pattern is used by many databases, such as Flybase.
+In the data, we only find references of bearers, such as anatomical entities or biological processes.
+Instead of explicitly stating phenotypic modifiers (abnormal, morphology, changed), it is implicit in the definition of the dataset.
+
+
+
+
dateAssigned
+
evidence/crossReference/id
+
evidence/publicationId
+
objectId
+
phenotypeStatement
+
phenotypeTermIdentifiers/0/termId
+
+
+
+
+
2024-01-05T11:54:24-05:00
+
FB:FBrf0052655
+
PMID:2385293
+
FB:FBal0016988
+
embryonic telson
+
FBbt:00000184
+
+
+
2024-01-05T11:54:24-05:00
+
FB:FBrf0058077
+
PMID:8223248
+
FB:FBal0001571
+
larva
+
FBbt:00001727
+
+
+
+
+
FBbt:00000184 (embryonic telson) and FBbt:00001727 (larva) are bearer terms.
Note: FlyBase has different kinds of phenotype data (including pre-coordinated), and it should be carefully evaluated which one is which prior to integration.
The most complex pattern for phenotype descriptions which essentially decomposes the entire phenotype expression into atomic consituents can be found, for example, in the The Zebrafish Information Network (ZFIN).
+
Examples:
+
+
+
+
Fish ID
+
Affected Structure or Process 1 subterm ID
+
Affected Structure or Process 1 subterm Name
+
Post-composed Relationship ID
+
Post-composed Relationship Name
+
Affected Structure or Process 1 superterm ID
+
Affected Structure or Process 1 superterm Name
+
Phenotype Keyword ID
+
Phenotype Keyword Name
+
Phenotype Tag
+
Affected Structure or Process 2 subterm ID
+
Affected Structure or Process 2 subterm name
+
Post-composed Relationship (rel) ID
+
Post-composed Relationship (rel) Name
+
Affected Structure or Process 2 superterm ID
+
Affected Structure or Process 2 superterm name
+
Publication ID
+
+
+
+
+
ZDB-FISH-150901-29105
+
ZFA:0009366
+
hair cell
+
BFO:0000050
+
part_of
+
ZFA:0000051
+
otic vesicle
+
PATO:0000374
+
increased distance
+
abnormal
+
ZFA:0009366
+
hair cell
+
BFO:0000050
+
part_of
+
ZFA:0000051
+
otic vesicle
+
ZDB-PUB-171025-12
+
+
+
ZDB-FISH-150901-29105
+
ZFA:0009366
+
hair cell
+
BFO:0000050
+
part_of
+
ZFA:0000051
+
otic vesicle
+
PATO:0000374
+
increased distance
+
abnormal
+
ZFA:0009366
+
hair cell
+
BFO:0000050
+
part_of
+
ZFA:0000051
+
otic vesicle
+
ZDB-PUB-171025-12
+
+
+
ZDB-FISH-150901-11537
+
+
+
+
+
ZFA:0000051
+
otic vesicle
+
PATO:0001905
+
has normal numbers of parts of type
+
normal
+
ZFA:0009366
+
hair cell
+
BFO:0000050
+
part_of
+
ZFA:0000051
+
otic vesicle
+
ZDB-PUB-150318-1
+
+
+
ZDB-FISH-150901-18770
+
+
+
+
+
ZFA:0000119
+
retinal inner nuclear layer
+
PATO:0002001
+
has fewer parts of type
+
abnormal
+
ZFA:0009315
+
horizontal cell
+
BFO:0000050
+
part_of
+
ZFA:0000119
+
retinal inner nuclear layer
+
ZDB-PUB-130222-28
+
+
+
ZDB-FISH-190806-7
+
BSPO:0000084
+
ventral region
+
BFO:0000050
+
part_of
+
ZFA:0000101
+
diencephalon
+
PATO:0002001
+
has fewer parts of type
+
abnormal
+
ZFA:0009301
+
dopaminergic neuron
+
BFO:0000050
+
part_of
+
ZFA:0000101
+
diencephalon
+
ZDB-PUB-190216-5
+
+
+
ZDB-FISH-190807-7
+
BSPO:0000084
+
ventral region
+
BFO:0000050
+
part_of
+
ZFA:0000101
+
diencephalon
+
PATO:0001905
+
has normal numbers of parts of type
+
normal
+
ZFA:0009301
+
dopaminergic neuron
+
BFO:0000050
+
part_of
+
ZFA:0000101
+
diencephalon
+
ZDB-PUB-190216-5
+
+
+
ZDB-FISH-190807-8
+
BSPO:0000084
+
ventral region
+
BFO:0000050
+
part_of
+
ZFA:0000101
+
diencephalon
+
PATO:0002001
+
has fewer parts of type
+
abnormal
+
ZFA:0009301
+
dopaminergic neuron
+
BFO:0000050
+
part_of
+
ZFA:0000101
+
diencephalon
+
ZDB-PUB-190216-5
+
+
+
ZDB-FISH-150901-29105
+
+
+
+
+
ZFA:0000101
+
diencephalon
+
PATO:0001555
+
has number of
+
normal
+
ZFA:0009301
+
dopaminergic neuron
+
BFO:0000050
+
part_of
+
ZFA:0000101
+
diencephalon
+
ZDB-PUB-161120-7
+
+
+
ZDB-FISH-210421-9
+
ZFA:0009290
+
glutamatergic neuron
+
BFO:0000050
+
part_of
+
ZFA:0000008
+
brain
+
PATO:0040043
+
increased proportionality to
+
abnormal
+
ZFA:0009276
+
GABAergic neuron
+
BFO:0000050
+
part_of
+
ZFA:0000008
+
brain
+
ZDB-PUB-191011-2
+
+
+
ZDB-FISH-210421-9
+
ZFA:0009290
+
glutamatergic neuron
+
BFO:0000050
+
part_of
+
ZFA:0000008
+
brain
+
PATO:0040043
+
increased proportionality to
+
abnormal
+
ZFA:0009276
+
GABAergic neuron
+
BFO:0000050
+
part_of
+
ZFA:0000008
+
brain
+
ZDB-PUB-191011-2
+
+
+
+
Lets break down the second to last row:
+
+
ZFA:0009290 (glutamatergic neuron): The primary entity whose characteristic is being observed
+
BFO:0000050 (part of): a relation used to connect the hair cell to the structure its part of
+
ZFA:0000008 (brain): the location of the primary entity being observed
+
PATO:0040043 (increased proportionality to): the modified characteristic being observed.
+
abnormal: the change modifier (note: not an ontology term)
+
ZFA:0009276 (GABAergic neuron): the secondary entity being observed in relation to which the characteristic is measured
+
ZFA:0000008 (brain): the location of the secondary entity
Phenotype data can be standardised to varying degrees. It is not uncommon for data to be completely unstandardised.
+Unfortunately, only a fraction of the available data is actually annotated using terms from controlled phenotype ontologies.
+Here are some of the more "typical" kinds of data on the standardised/non-standardised spectrum:
+
+
Free text in clinical notes and scientific publications
+
Free text in specific database fields (for example a "height" column in a table about measurements of Giraffes)
+
Controlled but non-standardised vocabulary like enums in a datamodel (for example the keyword "abnormal" in the ZFIN example above)
+
Controlled standardised vocabulary (like all the examples on this page)
+
Ontology terms (controlled vocabulary terms with well defined semantics - all the examples on this page)
Qualitative and quantitative phenotype data represent two fundamental ways of describing characteristics or traits in biology, each providing different types of information:
+
Qualitative Phenotype Data:
+
+
Nature: This type of data describes qualities or characteristics that are observed but not measured with numbers. It often involves categorical or descriptive information.
+
Examples: The presence or absence of a specific physical trait (like eye color or wing shape in animals) or types of behavior (aggressive vs. passive).
+
Analysis: Qualitative data is analyzed by categorization and identification of patterns or variations. It is more about the 'type' or 'kind' of trait rather than its 'amount'.
+
Interpretation: Since it's descriptive, this data relies on subjective interpretation and classification.
+
+
Quantitative Phenotype Data:
+
+
Nature: This data is numerical and quantifies traits. It involves measurements of characteristics, often allowing for more precise and objective analysis.
+
Examples: Height, weight, blood pressure, cholesterol levels, or the number of fruit produced by a plant. Quantitative traits can often be measured on a continuous scale, for example height of 35 cm, weight of 67 KG or blood pressure of 120/80.
+
Analysis: It involves statistical analysis, such as calculating mean, median, standard deviation, and applying various statistical tests. It allows for a more objective and replicable assessment.
+
Interpretation: Quantitative data provides a more concrete and measurable understanding of traits, making comparisons and statistical testing more straightforward.
+
+
Qualitative data is descriptive and categorical, while quantitative data is numerical and measurable. Both types are essential for a comprehensive understanding of phenotypic traits, each offering unique insights into biological variation and complexity.
Phenotype ontologies use different reference ontologies for their EQs. Everything in uPheno is integrated towards a common set of reference ontologies, in particular Uberon and CL. In order to integrate species-independent anatomy ontologies we employ the following workflow for phenotype ontologies:
+
+
Create a base-plus module from the ontology
+
Rename all Uberon-aligned entities using ROBOT rename. This replaces basically species specific anatomy references with Uberon anatomy references
+
Delete all species specific references from uPheno (FBBT, XAO, ZFA, etc). This also deletes all EQs which have non-Uberon references.
+
For all remaining species-specific anatomy terms, we retain only the link to the nearest Uberon term.
The uPheno project aims to unify the annotation of phenotypes across species in a manner analogous to unification of gene function annotation by the Gene Ontology. uPheno 2.0 builds on earlier efforts with a strategy that directly leverages the work of the phenotype ontology development community and incorporates phenotypes from a much wider range of species. We have organised a collaborative community effort, including representatives of all major model organism databases, to document and align formal design patterns for representing phenotypes and further develop reference ontologies, such as PATO, which are used in these patterns. A common development infrastructure makes it easy to use these design patterns to generate both species-specific ontologies and a species-independent layer that subsumes them. The resulting community-curated ontology for the representation and integration of phenotypes across species serves two general purposes: - Providing a community-developed framework for ontology editors to bootstrap, maintain and extend their phenotype ontologies in a scalable and standardised manner. - Facilitating the retrieval and comparative analysis of species-specific phenotypes through a deep layer of species-independent phenotypes.
Currently, the development of uPheno is organized by a group that meets biweekly. See the meetings page for more info, including how to participate.
"},{"location":"cite/","title":"How to cite uPheno","text":""},{"location":"cite/#papers","title":"Papers","text":""},{"location":"cite/#upheno-2","title":"uPheno 2","text":"
Matentzoglu N, Osumi-Sutherland D, Balhoff JP, Bello S, Bradford Y, Cardmody L, Grove C, Harris MA, Harris N, K\u00f6hler S, McMurry J, Mungall C, Munoz-Torres M, Pilgrim C, Robb S, Robinson PN, Segerdell E, Vasilevsky N, Haendel M. uPheno 2: Framework for standardised representation of phenotypes across species. 2019 Apr 8. http://dx.doi.org/10.7490/f1000research.1116540.1
Sebastian K\u00f6hler, Sandra C Doelken, Barbara J Ruef, Sebastian Bauer, Nicole Washington, Monte Westerfield, George Gkoutos, Paul Schofield, Damian Smedley, Suzanna E Lewis, Peter N Robinson, Christopher J Mungall (2013) Construction and accessibility of a cross-species phenotype ontology along with gene annotations for biomedical research F1000Research
"},{"location":"cite/#entity-quality-definitions-and-phenotype-modelling","title":"Entity-Quality definitions and phenotype modelling","text":"
C J Mungall, Georgios Gkoutos, Cynthia Smith, Melissa Haendel, Suzanna Lewis, Michael Ashburner (2010) Integrating phenotype ontologies across multiple species Genome Biology 11 (1)
"},{"location":"contributing/","title":"How to contribute to UPHENO","text":""},{"location":"howto/add-relation-extension/","title":"How to add the uPheno direct relation extension","text":"
EQ definitions are powerful tools for reconciling phenotypes across species and driving reasoning. However, they are not all that useful for many \"normal\" users of our ontologies.
We have developed a little workflow extension to take care of that.
As usual please follow the steps to install the custom uPheno Makefile extension first.
Now add a new component to your ont-odk.yaml file (e.g. src/ontology/mp-odk.yaml):
We can now choose if we want to add the component to your edit file as well. To do that, follow the instructions on adding an import (i.e. adding the component to the edit file and catalog file). The IRI of the component is http://purl.obolibrary.org/obo/YOURONTOLOGY/components/eq-relations.owl. For example, for MP, the IRI is http://purl.obolibrary.org/obo/mp/components/eq-relations.owl.
Now we can generate the component:
sh run.sh make components/eq-relations.owl\n
This command will be run automatically during a release (prepare_release).
The custom uPheno Makefile is an extension to your normal custom Makefile (for example, hp.Makefile, mp.Makefile, etc), located in the src/ontology directory of your ODK set up.
To install it:
(1) Open your normal custom Makefile and add a line in the very end:
Phenotype Ontology Working Group Meetings agenda and minutes gdoc.
phenotype-ontologies slack channel: to send meeting reminders; ask for agenda items; questions; discussions etc.
Dead simple owl design pattern (DOS-DP) Documentation
Getting started with DOSDP templates.
Dead Simple Ontology Design Patterns (DOSDP).
Using DOSDP templates in ODK Workflows.
Validate DOS-DP yaml templates:
yamllint: yaml syntax validator
Installing yamllint: brew install yamllint
Congiguring yamllint You can ignore the error line too long yaml syntax errors for dos-dp yaml templates. You can create a custom configuration file for yamllint in your home folder:
touch ~/.config/yamllint/config\n
The content of the config file should look like this:
# Custom configuration file for yamllint\n# It extends the default conf by adjusting some options.\nextends: default\nrules:\nline-length:\nmax: 80 # 80 chars should be enough, but don't fail if a line is longer\n# max: 140 # allow long lines\nlevel: warning\nallow-non-breakable-words: true\nallow-non-breakable-inline-mappings: true\n
The custom config should turn the error line too long errors to warnings.
DOS-DP validator:: DOS-DP format validator
Installing : pip install dosdp
Patternisation is the process of ensuring that all entity quality (EQ) descriptions from textual phenotype term definitions have a logical definition pattern. A pattern is a standard format for describing a phenotype that includes a quality and an entity. For example, \"increased body size\" is a pattern that includes the quality \"increased\" and the entity \"body size.\" The goal of patternisation is to make the EQ descriptions more uniform and machine-readable, which facilitates downstream analysis.
"},{"location":"howto/editors_workflow/#1-identify-a-group-of-related-phenotypes-from-diverse-organisms","title":"1. Identify a group of related phenotypes from diverse organisms","text":"
The first step in the Phenotype Ontology Editors' Workflow is to identify a group of related phenotypes from diverse organisms. This can be done by considering proposals from phenotype editors or by using the pattern suggestion pipeline. The phenotype editors may propose a group of related phenotypes based on their domain knowledge, while the pattern suggestion pipeline uses semantic similarity and shared Phenotype And Trait Ontology (PATO) quality terms to identify patterns in phenotype terms from different organism-specific ontologies.
"},{"location":"howto/editors_workflow/#2-propose-a-phenotype-pattern","title":"2. Propose a phenotype pattern","text":"
Once a group of related phenotypes is identified, the editors propose a phenotype pattern. To do this, they create a Github issue to request the phenotype pattern template in the uPheno repository. Alternatively, a new template can be proposed at a phenotype editors' meeting which can lead to the creation of a new term request as a Github issue. Ideally, the proposed phenotype pattern should include an appropriate PATO quality term for logical definition, use cases, term examples, and a textual definition pattern for the phenotype terms.
"},{"location":"howto/editors_workflow/#3-discuss-the-new-phenotype-pattern-draft-at-the-regular-upheno-phenotype-editors-meeting","title":"3. Discuss the new phenotype pattern draft at the regular uPheno phenotype editors meeting","text":"
The next step is to discuss the new phenotype pattern draft at the regular uPheno phenotype editors meeting. During the meeting, the editors' comments and suggestions for improvements are collected as comments on the DOS-DP yaml template in the corresponding Github pull request. Based on the feedback and discussions, a consensus on improvements should be achieved. The DOS-DP yaml template is named should start with a lower case letter, should be informative, and must include the PATO quality term. A Github pull request is created for the DOS-DP yaml template.
A DOS-DP phenotype pattern template example:
---\npattern_name: ??pattern_and_file_name\npattern_iri: http://purl.obolibrary.org/obo/upheno/patterns-dev/??pattern_and_file_name.yaml\ndescription: 'A description that helps people chose this pattern for the appropriate scenario.'\n# examples:\n# - example_IRI-1 # term name\n# - example_IRI-2 # term name\n# - example_IRI-3 # term name\n# - http://purl.obolibrary.org/obo/XXXXXXXXXX # XXXXXXXX\ncontributors:\n- https://orcid.org/XXXX-XXXX-XXXX-XXXX # Yyy Yyyyyyyyy\nclasses:\nprocess_quality: PATO:0001236\nabnormal: PATO:0000460\nanatomical_entity: UBERON:0001062\nrelations:\ncharacteristic_of: RO:0000052\nhas_modifier: RO:0002573\nhas_part: BFO:0000051\nannotationProperties:\nexact_synonym: oio:hasExactSynonym\nrelated_synonym: oio:hasRelatedSynonym\nxref: oio:hasDbXref\nvars:\nvar??: \"'anatomical_entity'\" # \"'variable_range'\"\nname:\ntext: \"trait ?? %s\"\nvars:\n- var??\nannotations:\n- annotationProperty: exact_synonym\ntext: \"? of %s\"\nvars:\n- var??\n- annotationProperty: related_synonym\ntext: \"? %s\"\nvars:\n- var??\n- annotationProperty: xref\ntext: \"AUTO:patterns/patterns/chemical_role_attribute\"\ndef:\ntext: \"A trait that ?? %s.\"\nvars:\n- var??\nequivalentTo:\ntext: \"'has_part' some (\n'XXXXXXXXXXXXXXXXX' and\n('characteristic_of' some %s) and\n('has_modifier' some 'abnormal')\n)\"\nvars:\n- var??\n...\n
"},{"location":"howto/editors_workflow/#4-review-the-candidate-phenotype-pattern","title":"4. Review the candidate phenotype pattern","text":"
Once a consensus on the improvements for a particular template is achieved, they are incorporated into the DOS-DP yaml file. Typically, the improvements are applied to the template some time before a subsequent ontology editor's meeting. There should be enough time for off-line review of the proposed pattern to allow community feedback. The improved phenotype pattern candidate draft should get approval from the community at one of the regular ontology editors' call or in a Github comment. The ontology editors who approve the pattern provide their ORCIDs and they are credited as contributors in an appropriate field of the DOS-DP pattern template.
"},{"location":"howto/editors_workflow/#5-add-the-community-approved-phenotype-pattern-template-to-upheno","title":"5. Add the community-approved phenotype pattern template to uPheno","text":"
Once the community-approved phenotype pattern template is created, it is added to the uPheno Github repository. The approved DOS-DP yaml phenotype pattern template should pass quality control (QC) steps. 1. Validate yaml syntax: yamllint 2. Validate DOS-DP Use DOSDP Validator. * To validate a template using the command line interface, execute: ```sh yamllint dosdp validate -i
After successfully passing QC, the responsible editor merges the approved pull request, and the phenotype pattern becomes part of the uPheno phenotype pattern template collection.
This document is on how to merge new DOSDP design patterns into an ODK ontology and then how to replace the old classes with the new ones.
"},{"location":"howto/pattern-merge-replace-workflow/#1-you-need-the-tables-in-tsv-format-with-the-dosdp-filler-data-download-the-tsv-tables-to","title":"1. You need the tables in tsv format with the DOSDP filler data. Download the tsv tables to","text":"
$ODK-ONTOLOGY/src/patterns/data/default/\n
Make sure that the tsv filenames match that of the relevant yaml DOSDP pattern files.
"},{"location":"howto/pattern-merge-replace-workflow/#2-add-the-new-matching-pattern-yaml-filename-to","title":"2. Add the new matching pattern yaml filename to","text":"
"},{"location":"howto/pattern-merge-replace-workflow/#3-import-the-new-pattern-templates-that-you-have-just-added-to-the-externaltxt-list-from-external-sources-into-the-current-working-repository","title":"3. Import the new pattern templates that you have just added to the external.txt list from external sources into the current working repository","text":"
cd ODK-ONTOLOGY/src/ontology\nsh run.sh make update_patterns\n
"},{"location":"howto/pattern-merge-replace-workflow/#4-make-definitionsowl","title":"4. make definitions.owl","text":"
cd ODK-ONTOLOGY/src/ontology\nsh run.sh make ../patterns/definitions.owl IMP=false\n
"},{"location":"howto/pattern-merge-replace-workflow/#5-remove-old-classes-and-replace-them-with-the-equivalent-and-patternised-new-classes","title":"5. Remove old classes and replace them with the equivalent and patternised new classes","text":"
cd ODK-ONTOLOGY/src/ontology\nsh run.sh make remove_patternised_classes\n
"},{"location":"howto/pattern-merge-replace-workflow/#6-announce-the-pattern-migration-in-an-appropriate-channel-for-example-on-the-phenotype-ontologies-slack-channel","title":"6. Announce the pattern migration in an appropriate channel, for example on the phenotype-ontologies Slack channel.","text":"
For example:
I have migrated the ... table and changed the tab colour to blue. You can delete the tab if you wish.
"},{"location":"howto/run-upheno2-release/","title":"How to run a uPheno 2 release","text":"
In order to run a release you will have to have completed the steps to set up s3.
Clone https://github.com/obophenotype/upheno-dev
cd src/scripts
sh upheno_pipeline.sh
cd ../ontology
make prepare_upload S3_VERSION=2022-06-19
make deploy S3_VERSION=2022-06-19
"},{"location":"howto/set-up-s3/","title":"How to set yourself up for S3","text":"
To be able to upload new uPheno release to the uPheno S3 bucket, you need to set yourself up for S3 first.
Download and install AWS CLI
Obtain secrets from BBOP
Add configuration for secrets
"},{"location":"howto/set-up-s3/#1-download-and-install-aws-cli","title":"1. Download and install AWS CLI","text":"
The most convenient way to interact with S3 is the AWS Command Line Interface (CLI). You can find the installers and install instructions on that page (different depending on your Operation System): - For Mac - For Windows
"},{"location":"howto/set-up-s3/#2-obtain-secrets-from-bbop","title":"2. Obtain secrets from BBOP","text":"
Next, you need to ask someone at BBOP (such as Chris Mungall or Seth Carbon) to provide you with an account that gives you access to the BBOP s3 buckets. You will have to provide a username. You will receive: - User name - Access key ID- - Secret access key - Console link to sign into bucket
"},{"location":"howto/set-up-s3/#3-add-configuration-for-secrets","title":"3. Add configuration for secrets","text":"
You will now have to set up your local system. You will create two files:
$ less ~/.aws/config \n[default]\nregion = us-east-1\n
and
$ less ~/.aws/credentials\n[default]\naws_access_key_id = ***\naws_secret_access_key = ***\n
in ~/.aws/credentials make sure you add the correct keys as provided above.
"},{"location":"howto/set-up-s3/#4-write-to-your-bucket","title":"4. Write to your bucket","text":"
Now, you should be set up to write to your s3 bucket. Note that in order for your data to be accessible through https after your upload, you need to add --acl public read.
If you have previously pushed data to the same location, you wont be able to set it to \"publicly readable\" by simply rerunning the sync command. If you want to publish previously private data, follow the instructions here, e.g.:
"},{"location":"odk-workflows/ContinuousIntegration/","title":"Introduction to Continuous Integration Workflows with ODK","text":"
Historically, most repos have been using Travis CI for continuous integration testing and building, but due to runtime restrictions, we recently switched a lot of our repos to GitHub actions. You can set up your repo with CI by adding this to your configuration file (src/ontology/upheno-odk.yaml):
ci:\n - github_actions\n
When updateing your repo, you will notice a new file being added: .github/workflows/qc.yml.
This file contains your CI logic, so if you need to change, or add anything, this is the place!
Alternatively, if your repo is in GitLab instead of GitHub, you can set up your repo with GitLab CI by adding this to your configuration file (src/ontology/upheno-odk.yaml):
ci:\n - gitlab-ci\n
This will add a file called .gitlab-ci.yml in the root of your repo.
The editors workflow is one of the formal workflows to ensure that the ontology is developed correctly according to ontology engineering principles. There are a few different editors workflows:
Local editing workflow: Editing the ontology in your local environment by hand, using tools such as Prot\u00e9g\u00e9, ROBOT templates or DOSDP patterns.
Completely automated data pipeline (GitHub Actions)
DROID workflow
This document only covers the first editing workflow, but more will be added in the future
Ensure that there is a ticket on your issue tracker that describes the change you are about to make. While this seems optional, this is a very important part of the social contract of building an ontology - no change to the ontology should be performed without a good ticket, describing the motivation and nature of the intended change.
"},{"location":"odk-workflows/EditorsWorkflow/#2-update-main-branch","title":"2. Update main branch","text":"
In your local environment (e.g. your laptop), make sure you are on the main (prev. master) branch and ensure that you have all the upstream changes, for example:
Create a new branch. Per convention, we try to use meaningful branch names such as: - issue23removeprocess (where issue 23 is the related issue on GitHub) - issue26addcontributor - release20210101 (for releases)
Using your editor of choice, perform the intended edit. For example:
Prot\u00e9g\u00e9
Open src/ontology/upheno-edit.owl in Prot\u00e9g\u00e9
Make the change
Save the file
TextEdit
Open src/ontology/upheno-edit.owl in TextEdit (or Sublime, Atom, Vim, Nano)
Make the change
Save the file
Consider the following when making the edit.
According to our development philosophy, the only places that should be manually edited are:
src/ontology/upheno-edit.owl
Any ROBOT templates you chose to use (the TSV files only)
Any DOSDP data tables you chose to use (the TSV files, and potentially the associated patterns)
components (anything in src/ontology/components), see here.
Imports should not be edited (any edits will be flushed out with the next update). However, refreshing imports is a potentially breaking change - and is discussed elsewhere.
Changes should usually be small. Adding or changing 1 term is great. Adding or changing 10 related terms is ok. Adding or changing 100 or more terms at once should be considered very carefully.
"},{"location":"odk-workflows/EditorsWorkflow/#4-check-the-git-diff","title":"4. Check the Git diff","text":"
This step is very important. Rather than simply trusting your change had the intended effect, we should always use a git diff as a first pass for sanity checking.
In our experience, having a visual git client like GitHub Desktop or sourcetree is really helpful for this part. In case you prefer the command line:
Now it's time to run your quality control checks. This can either happen locally (5a) or through your continuous integration system (7/5b).
"},{"location":"odk-workflows/EditorsWorkflow/#5a-local-testing","title":"5a. Local testing","text":"
If you chose to run your test locally:
sh run.sh make IMP=false test\n
This will run the whole set of configured ODK tests on including your change. If you have a complex DOSDP pattern pipeline you may want to add PAT=false to skip the potentially lengthy process of rebuilding the patterns.
When you are happy with the changes, you commit your changes to your feature branch, push them upstream (to GitHub) and create a pull request. For example:
git add NAMEOFCHANGEDFILES\ngit commit -m \"Added biological process term #12\"\ngit push -u origin issue23removeprocess\n
Then you go to your project on GitHub, and create a new pull request from the branch, for example: https://github.com/INCATools/ontology-development-kit/pulls
There is a lot of great advise on how to write pull requests, but at the very least you should: - mention the tickets affected: see #23 to link to a related ticket, or fixes #23 if, by merging this pull request, the ticket is fixed. Tickets in the latter case will be closed automatically by GitHub when the pull request is merged. - summarise the changes in a few sentences. Consider the reviewer: what would they want to know right away. - If the diff is large, provide instructions on how to review the pull request best (sometimes, there are many changed files, but only one important change).
If you didn't run and local quality control checks (see 5a), you should have Continuous Integration (CI) set up, for example: - Travis - GitHub Actions
More on how to set this up here. Once the pull request is created, the CI will automatically trigger. If all is fine, it will show up green, otherwise red.
"},{"location":"odk-workflows/EditorsWorkflow/#8-community-review","title":"8. Community review","text":"
Once all the automatic tests have passed, it is important to put a second set of eyes on the pull request. Ontologies are inherently social - as in that they represent some kind of community consensus on how a domain is organised conceptually. This seems high brow talk, but it is very important that as an ontology editor, you have your work validated by the community you are trying to serve (e.g. your colleagues, other contributors etc.). In our experience, it is hard to get more than one review on a pull request - two is great. You can set up GitHub branch protection to actually require a review before a pull request can be merged! We recommend this.
This step seems daunting to some hopefully under-resourced ontologies, but we recommend to put this high up on your list of priorities - train a colleague, reach out!
"},{"location":"odk-workflows/EditorsWorkflow/#9-merge-and-cleanup","title":"9. Merge and cleanup","text":"
When the QC is green and the reviews are in (approvals), it is time to merge the pull request. After the pull request is merged, remember to delete the branch as well (this option will show up as a big button right after you have merged the pull request). If you have not done so, close all the associated tickets fixed by the pull request.
It is sometimes difficult to keep track of changes made to an ontology. Some ontology teams opt to document changes in a changelog (simply a text file in your repository) so that when release day comes, you know everything you have changed. This is advisable at least for major changes (such as a new release system, a new pattern or template etc.).
We can define custom checks using SPARQL. SPARQL queries define bad modelling patterns (missing labels, misspelt URIs, and many more) in the ontology. If these queries return any results, then the build will fail. Custom checks are designed to be run as part of GitHub Actions Continuous Integration testing, but they can also run locally.
"},{"location":"odk-workflows/ManageAutomatedTest/#steps-to-add-a-constraint-violation-check","title":"Steps to add a constraint violation check:","text":"
Add the SPARQL query in src/sparql. The name of the file should end with -violation.sparql. Please give a name that helps to understand which violation the query wants to check.
Add the name of the new file to odk configuration file src/ontology/uberon-odk.yaml:
Include the name of the file (without the -violation.sparql part) to the list inside the key custom_sparql_checks that is inside robot_report key.
If the robot_report or custom_sparql_checks keys are not available, please add this code block to the end of the file.
3. Update the repository so your new SPARQL check will be included in the QC.
sh run.sh make update_repo\n
"},{"location":"odk-workflows/ManageDocumentation/","title":"Updating the Documentation","text":"
The documentation for UPHENO is managed in two places (relative to the repository root):
The docs directory contains all the files that pertain to the content of the documentation (more below)
the mkdocs.yaml file contains the documentation config, in particular its navigation bar and theme.
The documentation is hosted using GitHub pages, on a special branch of the repository (called gh-pages). It is important that this branch is never deleted - it contains all the files GitHub pages needs to render and deploy the site. It is also important to note that the gh-pages branch should never be edited manually. All changes to the docs happen inside the docs directory on the main branch.
"},{"location":"odk-workflows/ManageDocumentation/#editing-the-docs","title":"Editing the docs","text":""},{"location":"odk-workflows/ManageDocumentation/#changing-content","title":"Changing content","text":"
All the documentation is contained in the docs directory, and is managed in Markdown. Markdown is a very simple and convenient way to produce text documents with formatting instructions, and is very easy to learn - it is also used, for example, in GitHub issues. This is a normal editing workflow:
Open the .md file you want to change in an editor of choice (a simple text editor is often best). IMPORTANT: Do not edit any files in the docs/odk-workflows/ directory. These files are managed by the ODK system and will be overwritten when the repository is upgraded! If you wish to change these files, make an issue on the ODK issue tracker.
Perform the edit and save the file
Commit the file to a branch, and create a pull request as usual.
If your development team likes your changes, merge the docs into master branch.
Deploy the documentation (see below)
"},{"location":"odk-workflows/ManageDocumentation/#deploy-the-documentation","title":"Deploy the documentation","text":"
The documentation is not automatically updated from the Markdown, and needs to be deployed deliberately. To do this, perform the following steps:
In your terminal, navigate to the edit directory of your ontology, e.g.:
cd upheno/src/ontology\n
Now you are ready to build the docs as follows:
sh run.sh make update_docs\n
Mkdocs now sets off to build the site from the markdown pages. You will be asked to
Enter your username
Enter your password (see here for using GitHub access tokens instead) IMPORTANT: Using password based authentication will be deprecated this year (2021). Make sure you read up on personal access tokens if that happens!
If everything was successful, you will see a message similar to this one:
INFO - Your documentation should shortly be available at: https://obophenotype.github.io/upheno/ \n
3. Just to double check, you can now navigate to your documentation pages (usually https://obophenotype.github.io/upheno/). Just make sure you give GitHub 2-5 minutes to build the pages!"},{"location":"odk-workflows/ReleaseWorkflow/","title":"The release workflow","text":"
The release workflow recommended by the ODK is based on GitHub releases and works as follows:
Run a release with the ODK
Review the release
Merge to main branch
Create a GitHub release
These steps are outlined in detail in the following.
"},{"location":"odk-workflows/ReleaseWorkflow/#run-a-release-with-the-odk","title":"Run a release with the ODK","text":"
Preparation:
Ensure that all your pull requests are merged into your main (master) branch
Make sure that all changes to master are committed to GitHub (git status should say that there are no modified files)
Locally make sure you have the latest changes from master (git pull)
Checkout a new branch (e.g. git checkout -b release-2021-01-01)
You may or may not want to refresh your imports as part of your release strategy (see here)
Make sure you have the latest ODK installed by running docker pull obolibrary/odkfull
To actually run the release, you:
Open a command line terminal window and navigate to the src/ontology directory (cd upheno/src/ontology)
Run release pipeline:sh run.sh make prepare_release -B. Note that for some ontologies, this process can take up to 90 minutes - especially if there are large ontologies you depend on, like PRO or CHEBI.
If everything went well, you should see the following output on your machine: Release files are now in ../.. - now you should commit, push and make a release on your git hosting site such as GitHub or GitLab.
This will create all the specified release targets (OBO, OWL, JSON, and the variants, ont-full and ont-base) and copy them into your release directory (the top level of your repo).
"},{"location":"odk-workflows/ReleaseWorkflow/#review-the-release","title":"Review the release","text":"
(Optional) Rough check. This step is frequently skipped, but for the more paranoid among us (like the author of this doc), this is a 3 minute additional effort for some peace of mind. Open the main release (upheno.owl) in you favourite development environment (i.e. Prot\u00e9g\u00e9) and eyeball the hierarchy. We recommend two simple checks:
Does the very top level of the hierarchy look ok? This means that all new terms have been imported/updated correctly.
Does at least one change that you know should be in this release appear? For example, a new class. This means that the release was actually based on the recent edit file.
Commit your changes to the branch and make a pull request
In your GitHub pull request, review the following three files in detail (based on our experience):
upheno.obo - this reflects a useful subset of the whole ontology (everything that can be covered by OBO format). OBO format has that speaking for it: it is very easy to review!
upheno-base.owl - this reflects the asserted axioms in your ontology that you have actually edited.
Ideally also take a look at upheno-full.owl, which may reveal interesting new inferences you did not know about. Note that the diff of this file is sometimes quite large.
Like with every pull request, we recommend to always employ a second set of eyes when reviewing a PR!
"},{"location":"odk-workflows/ReleaseWorkflow/#merge-the-main-branch","title":"Merge the main branch","text":"
Once your CI checks have passed, and your reviews are completed, you can now merge the branch into your main branch (don't forget to delete the branch afterwards - a big button will appear after the merge is finished).
"},{"location":"odk-workflows/ReleaseWorkflow/#create-a-github-release","title":"Create a GitHub release","text":"
Go to your releases page on GitHub by navigating to your repository, and then clicking on releases (usually on the right, for example: https://github.com/obophenotype/upheno/releases). Then click \"Draft new release\"
As the tag version you need to choose the date on which your ontologies were build. You can find this, for example, by looking at the upheno.obo file and check the data-version: property. The date needs to be prefixed with a v, so, for example v2020-02-06.
You can write whatever you want in the release title, but we typically write the date again. The description underneath should contain a concise list of changes or term additions.
Click \"Publish release\". Done.
"},{"location":"odk-workflows/ReleaseWorkflow/#debugging-typical-ontology-release-problems","title":"Debugging typical ontology release problems","text":""},{"location":"odk-workflows/ReleaseWorkflow/#problems-with-memory","title":"Problems with memory","text":"
When you are dealing with large ontologies, you need a lot of memory. When you see error messages relating to large ontologies such as CHEBI, PRO, NCBITAXON, or Uberon, you should think of memory first, see here.
"},{"location":"odk-workflows/ReleaseWorkflow/#problems-when-using-obo-format-based-tools","title":"Problems when using OBO format based tools","text":"
Sometimes you will get cryptic error messages when using legacy tools using OBO format, such as the ontology release tool (OORT), which is also available as part of the ODK docker container. In these cases, you need to track down what axiom or annotation actually caused the breakdown. In our experience (in about 60% of the cases) the problem lies with duplicate annotations (def, comment) which are illegal in OBO. Here is an example recipe of how to deal with such a problem:
If you get a message like make: *** [cl.Makefile:84: oort] Error 255 you might have a OORT error.
To debug this, in your terminal enter sh run.sh make IMP=false PAT=false oort -B (assuming you are already in the ontology folder in your directory)
This should show you where the error is in the log (eg multiple different definitions) WARNING: THE FIX BELOW IS NOT IDEAL, YOU SHOULD ALWAYS TRY TO FIX UPSTREAM IF POSSIBLE
Open upheno-edit.owl in Prot\u00e9g\u00e9 and find the offending term and delete all offending issue (e.g. delete ALL definition, if the problem was \"multiple def tags not allowed\") and save. *While this is not idea, as it will remove all definitions from that term, it will be added back again when the term is fixed in the ontology it was imported from and added back in.
Rerun sh run.sh make IMP=false PAT=false oort -B and if it all passes, commit your changes to a branch and make a pull request as usual.
"},{"location":"odk-workflows/RepoManagement/","title":"Managing your ODK repository","text":""},{"location":"odk-workflows/RepoManagement/#updating-your-odk-repository","title":"Updating your ODK repository","text":"
Your ODK repositories configuration is managed in src/ontology/upheno-odk.yaml. Once you have made your changes, you can run the following to apply your changes to the repository:
sh run.sh make update_repo\n
There are a large number of options that can be set to configure your ODK, but we will only discuss a few of them here.
NOTE for Windows users:
You may get a cryptic failure such as Set Illegal Option - if the update script located in src/scripts/update_repo.sh was saved using Windows Line endings. These need to change to unix line endings. In Notepad++, for example, you can click on Edit->EOL Conversion->Unix LF to change this.
You can use the update repository workflow described on this page to perform the following operations to your imports:
Add a new import
Modify an existing import
Remove an import you no longer want
Customise an import
We will discuss all these workflows in the following.
"},{"location":"odk-workflows/RepoManagement/#add-new-import","title":"Add new import","text":"
To add a new import, you first edit your odk config as described above, adding an id to the product list in the import_group section (for the sake of this example, we assume you already import RO, and your goal is to also import GO):
import_group:\n products:\n - id: ro\n - id: go\n
Note: our ODK file should only have one import_group which can contain multiple imports (in the products section). Next, you run the update repo workflow to apply these changes. Note that by default, this module is going to be a SLME Bottom module, see here. To change that or customise your module, see section \"Customise an import\". To finalise the addition of your import, perform the following steps:
Add an import statement to your src/ontology/upheno-edit.owl file. We suggest to do this using a text editor, by simply copying an existing import declaration and renaming it to the new ontology import, for example as follows:
in your catalog, tools like robot or Prot\u00e9g\u00e9 will recognize the statement in the catalog file to redirect the URL http://purl.obolibrary.org/obo/upheno/imports/go_import.owl to the local file imports/go_import.owl (which is in your src/ontology directory).
"},{"location":"odk-workflows/RepoManagement/#modify-an-existing-import","title":"Modify an existing import","text":"
If you simply wish to refresh your import in light of new terms, see here. If you wish to change the type of your module see section \"Customise an import\".
"},{"location":"odk-workflows/RepoManagement/#remove-an-existing-import","title":"Remove an existing import","text":"
To remove an existing import, perform the following steps:
remove the import declaration from your src/ontology/upheno-edit.owl.
remove the id from your src/ontology/upheno-odk.yaml, eg. - id: go from the list of products in the import_group.
run update repo workflow
delete the associated files manually:
src/imports/go_import.owl
src/imports/go_terms.txt
Remove the respective entry from the src/ontology/catalog-v001.xml file.
"},{"location":"odk-workflows/RepoManagement/#customise-an-import","title":"Customise an import","text":"
By default, an import module extracted from a source ontology will be a SLME module, see here. There are various options to change the default.
The following change to your repo config (src/ontology/upheno-odk.yaml) will switch the go import from an SLME module to a simple ROBOT filter module:
A ROBOT filter module is, essentially, importing all external terms declared by your ontology (see here on how to declare external terms to be imported). Note that the filter module does not consider terms/annotations from namespaces other than the base-namespace of the ontology itself. For example, in the example of GO above, only annotations / axioms related to the GO base IRI (http://purl.obolibrary.org/obo/GO_) would be considered. This behaviour can be changed by adding additional base IRIs as follows:
If you wish to customise your import entirely, you can specify your own ROBOT command to do so. To do that, add the following to your repo config (src/ontology/upheno-odk.yaml):
Now feel free to change this goal to do whatever you wish it to do! It probably makes some sense (albeit not being a strict necessity), to leave most of the goal instead and replace only:
When running sh run.sh make update_repo, a new file src/ontology/components/mycomp.owl will be created which you can edit as you see fit. Typical ways to edit:
Using a ROBOT template to generate the component (see below)
Manually curating the component separately with Prot\u00e9g\u00e9 or any other editor
Providing a components/mycomp.owl: make target in src/ontology/upheno.Makefile and provide a custom command to generate the component
WARNING: Note that the custom rule to generate the component MUST NOT depend on any other ODK-generated file such as seed files and the like (see issue).
Providing an additional attribute for the component in src/ontology/upheno-odk.yaml, source, to specify that this component should simply be downloaded from somewhere on the web.
"},{"location":"odk-workflows/RepoManagement/#adding-a-new-component-based-on-a-robot-template","title":"Adding a new component based on a ROBOT template","text":"
Since ODK 1.3.2, it is possible to simply link a ROBOT template to a component without having to specify any of the import logic. In order to add a new component that is connected to one or more template files, follow these steps:
Open src/ontology/upheno-odk.yaml.
Make sure that use_templates: TRUE is set in the global project options. You should also make sure that use_context: TRUE is set in case you are using prefixes in your templates that are not known to robot, such as OMOP:, CPONT: and more. All non-standard prefixes you are using should be added to config/context.json.
Add another component to the products section.
To activate this component to be template-driven, simply say: use_template: TRUE. This will create an empty template for you in the templates directory, which will automatically be processed when recreating the component (e.g. run.bat make recreate-mycomp).
If you want to use more than one component, use the templates field to add as many template names as you wish. ODK will look for them in the src/templates directory.
Advanced: If you want to provide additional processing options, you can use the template_options field. This should be a string with option from robot template. One typical example for additional options you may want to provide is --add-prefixes config/context.json to ensure the prefix map of your context is provided to robot, see above.
Release file are the file that are considered part of the official ontology release and to be used by the community. A detailed description of the release artefacts can be found here.
Imports are subsets of external ontologies that contain terms and axioms you would like to re-use in your ontology. These are considered \"external\", like dependencies in software development, and are not included in your \"base\" product, which is the release artefact which contains only those axioms that you personally maintain.
Components, in contrast to imports, are considered full members of the ontology. This means that any axiom in a component is also included in the ontology base - which means it is considered native to the ontology. While this sounds complicated, consider this: conceptually, no component should be part of more than one ontology. If that seems to be the case, we are most likely talking about an import. Components are often not needed for ontologies, but there are some use cases:
There is an automated process that generates and re-generates a part of the ontology
A part of the ontology is managed in ROBOT templates
The expressivity of the component is higher than the format of the edit file. For example, people still choose to manage their ontology in OBO format (they should not) missing out on a lot of owl features. They may choose to manage logic that is beyond OBO in a specific OWL component.
These are the components in UPHENO
Filename URL phenotypes_manual.owl None upheno-mappings.owl None cross-species-mappings.owl None"},{"location":"odk-workflows/SettingUpDockerForODK/","title":"Setting up your Docker environment for ODK use","text":"
One of the most frequent problems with running the ODK for the first time is failure because of lack of memory. This can look like a Java OutOfMemory exception, but more often than not it will appear as something like an Error 137. There are two places you need to consider to set your memory:
Your src/ontology/run.sh (or run.bat) file. You can set the memory in there by adding robot_java_args: '-Xmx8G' to your src/ontology/upheno-odk.yaml file, see for example here.
Set your docker memory. By default, it should be about 10-20% more than your robot_java_args variable. You can manage your memory settings by right-clicking on the docker whale in your system bar-->Preferences-->Resources-->Advanced, see picture below.
This page discusses how to update the contents of your imports, like adding or removing terms. If you are looking to customise imports, like changing the module type, see here.
"},{"location":"odk-workflows/UpdateImports/#importing-a-new-term","title":"Importing a new term","text":"
Note: some ontologies now use a merged-import system to manage dynamic imports, for these please follow instructions in the section title \"Using the Base Module approach\".
Importing a new term is split into two sub-phases:
Declaring the terms to be imported
Refreshing imports dynamically
"},{"location":"odk-workflows/UpdateImports/#declaring-terms-to-be-imported","title":"Declaring terms to be imported","text":"
There are three ways to declare terms that are to be imported from an external ontology. Choose the appropriate one for your particular scenario (all three can be used in parallel if need be):
This workflow is to be avoided, but may be appropriate if the editor does not have access to the ODK docker container. This approach also applies to ontologies that use base module import approach.
Open your ontology (edit file) in Prot\u00e9g\u00e9 (5.5+).
Select 'owl:Thing'
Add a new class as usual.
Paste the full iri in the 'Name:' field, for example, http://purl.obolibrary.org/obo/CHEBI_50906.
Click 'OK'
Now you can use this term for example to construct logical definitions. The next time the imports are refreshed (see how to refresh here), the metadata (labels, definitions, etc.) for this term are imported from the respective external source ontology and becomes visible in your ontology.
"},{"location":"odk-workflows/UpdateImports/#using-term-files","title":"Using term files","text":"
Every import has, by default a term file associated with it, which can be found in the imports directory. For example, if you have a GO import in src/ontology/go_import.owl, you will also have an associated term file src/ontology/go_terms.txt. You can add terms in there simply as a list:
GO:0008150\nGO:0008151\n
Now you can run the refresh imports workflow) and the two terms will be imported.
"},{"location":"odk-workflows/UpdateImports/#using-the-custom-import-template","title":"Using the custom import template","text":"
This workflow is appropriate if:
You prefer to manage all your imported terms in a single file (rather than multiple files like in the \"Using term files\" workflow above).
You wish to augment your imported ontologies with additional information. This requires a cautionary discussion.
To enable this workflow, you add the following to your ODK config file (src/ontology/upheno-odk.yaml), and update the repository:
use_custom_import_module: TRUE\n
Now you can manage your imported terms directly in the custom external terms template, which is located at src/templates/external_import.owl. Note that this file is a ROBOT template, and can, in principle, be extended to include any axioms you like. Before extending the template, however, read the following carefully.
The main purpose of the custom import template is to enable the management off all terms to be imported in a centralised place. To enable that, you do not have to do anything other than maintaining the template. So if you, say currently import APOLLO_SV:00000480, and you wish to import APOLLO_SV:00000532, you simply add a row like this:
ID Entity Type\nID TYPE\nAPOLLO_SV:00000480 owl:Class\nAPOLLO_SV:00000532 owl:Class\n
When the imports are refreshed see imports refresh workflow, the term(s) will simply be imported from the configured ontologies.
Now, if you wish to extend the Makefile (which is beyond these instructions) and add, say, synonyms to the imported terms, you can do that, but you need to (a) preserve the ID and ENTITY columns and (b) ensure that the ROBOT template is valid otherwise, see here.
WARNING. Note that doing this is a widespread antipattern (see related issue). You should not change the axioms of terms that do not belong into your ontology unless necessary - such changes should always be pushed into the ontology where they belong. However, since people are doing it, whether the OBO Foundry likes it or not, at least using the custom imports module as described here localises the changes to a single simple template and ensures that none of the annotations added this way are merged into the base file.
If you want to refresh the import yourself (this may be necessary to pass the travis tests), and you have the ODK installed, you can do the following (using go as an example):
First, you navigate in your terminal to the ontology directory (underneath src in your hpo root directory).
cd src/ontology\n
Then, you regenerate the import that will now include any new terms you have added. Note: You must have docker installed.
sh run.sh make PAT=false imports/go_import.owl -B\n
Since ODK 1.2.27, it is also possible to simply run the following, which is the same as the above:
sh run.sh make refresh-go\n
Note that in case you changed the defaults, you need to add IMP=true and/or MIR=true to the command below:
sh run.sh make IMP=true MIR=true PAT=false imports/go_import.owl -B\n
If you wish to skip refreshing the mirror, i.e. skip downloading the latest version of the source ontology for your import (e.g. go.owl for your go import) you can set MIR=false instead, which will do the exact same thing as the above, but is easier to remember:
sh run.sh make IMP=true MIR=false PAT=false imports/go_import.owl -B\n
"},{"location":"odk-workflows/UpdateImports/#using-the-base-module-approach","title":"Using the Base Module approach","text":"
Since ODK 1.2.31, we support an entirely new approach to generate modules: Using base files. The idea is to only import axioms from ontologies that actually belong to it. A base file is a subset of the ontology that only contains those axioms that nominally belong there. In other words, the base file does not contain any axioms that belong to another ontology. An example would be this:
The base file pipeline is a bit more complex than the normal pipelines, because of the logical interactions between the imported ontologies. This is solved by _first merging all mirrors into one huge file and then extracting one mega module from it.
Example: Let's say we are importing terms from Uberon, GO and RO in our ontologies. When we use the base pipelines, we
1) First obtain the base (usually by simply downloading it, but there is also an option now to create it with ROBOT) 2) We merge all base files into one big pile 3) Then we extract a single module imports/merged_import.owl
The first implementation of this pipeline is PATO, see https://github.com/pato-ontology/pato/blob/master/src/ontology/pato-odk.yaml.
To check if your ontology uses this method, check src/ontology/upheno-odk.yaml to see if use_base_merging: TRUE is declared under import_group
If your ontology uses Base Module approach, please use the following steps:
First, add the term to be imported to the term file associated with it (see above \"Using term files\" section if this is not clear to you)
Next, you navigate in your terminal to the ontology directory (underneath src in your hpo root directory).
cd src/ontology\n
Then refresh imports by running
sh run.sh make imports/merged_import.owl\n
Note: if your mirrors are updated, you can run sh run.sh make no-mirror-refresh-merged
This requires quite a bit of memory on your local machine, so if you encounter an error, it might be a lack of memory on your computer. A solution would be to create a ticket in an issue tracker requesting for the term to be imported, and one of the local devs should pick this up and run the import for you.
Lastly, restart Prot\u00e9g\u00e9, and the term should be imported in ready to be used.
"},{"location":"odk-workflows/components/","title":"Adding components to an ODK repo","text":"
For details on what components are, please see component section of repository file structure document.
To add custom components to an ODK repo, please follow the following steps:
1) Locate your odk yaml file and open it with your favourite text editor (src/ontology/upheno-odk.yaml) 2) Search if there is already a component section to the yaml file, if not add it accordingly, adding the name of your component:
5) Refresh your repo by running sh run.sh make update_repo - this should create a new file in src/ontology/components. 6) In your custom makefile (src/ontology/upheno.Makefile) add a goal for your custom make file. In this example, the goal is a ROBOT template.
The uPheno editors call is held every second Thursday (bi-weekly) on Zoom, provided by members of the Monarch Initiative and co-organised by members of the Alliance and Genome Resources. If you wish to join the meeting, you can open an issue on https://github.com/obophenotype/upheno/issues with the request to be added, or send an email to phenotype-ontologies-editors@googlegroups.com.
The meeting coordinator (MC) is the person charged with organising the meeting. The current MC is Ray, @rays22.
The MC prepares the agenda in advance: everyone on the call is very busy and our time is precious.
Every agenda item has an associated ticket on GitHub, and a clear set of action items should be added in GitHub Tasklist syntax to the first comment on the issue.
If there are issues for any subtasks (e.g. PATO or Uberon edits), the list should be edited to link these.
Any items that do not have a subissue but do involve changes to patterns) should be edited to link to implementing PR.
It does not matter who wrote the first issue comment, the uPheno team can simply add a tasklist underneath the original comment and refine it over time.
Tag all issues which need discussion with \"upheno call\"
It must be clear from the task list what the uPheno team should be doing during the call (discuss, decide, review). For example, one item on the task list may read: \"uPheno team to decide on appropriate label for template\".
Conversely, no issue should be added to the agenda that does not have a clear set of action items associated with it that should be addressed during the call. These actions may include making and documenting modelling decisions.
Go through up to 10 issues on the uPheno issue tracker before each meeting to determine how to progress on them, and add action items. Only if they need to be discussed, add the \"upheno call\" label.
Every meeting should start with a quick (max 5 min, ideally 3 min) overview of all the goals and how they processed. The MC should mention all blockers and goals, even the ones we did not make any progress on, to keep the focus on the priorities:
uPheno releases
uPheno documentation
Pattern creation
Patternisation: The process of ensuring that phenotype ontologies are using uPheno conformant templates to define their phenotypes.
Harmonisation: The process of ensuring that phenotype patterns are applied consistently across ontologies.
For new pattern discussions:
Every new pattern proposal should come with a new GitHub issue, appropriately tagged.
The issue text should detail the use cases for the pattern well, and these use cases should also be documented in the \"description\" part of the DOSDP YAML file. Uses cases should include expected classifications and why we want them (and potentially classifications to avoid). e.g. axis-specific dimension traits should classify under more abstractly defined dimension traits which in term should classify under Morphology. Add some examples of contexts where grouping along these classifications is useful.
Agenda items may include discussion and decisions about more general modelling issues that affect more than one pattern, but these should also be documented as tickets as described above.
"},{"location":"organization/meetings/#after-the-meeting","title":"After the meeting","text":"
After every meeting, update all issues discussed on GitHub and, in particular, clarify the remaining action items.
Ensure that the highest priority issues are discussed first.
"},{"location":"organization/outreach/","title":"The Outreach Programme of the Unified Phenotype Ontology (uPheno) development team","text":""},{"location":"organization/outreach/#outreach-calls","title":"Outreach-calls","text":"
The uPheno organises an outreach call every four weeks to listen to external stakeholders describing their need for cross-species phenotype integration.
"},{"location":"organization/outreach/#schedule","title":"Schedule","text":"Date Lesson Notes Recordings 2024/04/05 TBD TBD 2024/3/08 Computational identification of disease models through cross-species phenotype comparison Diego A. Pava, Pilar Cacheiro, Damian Smedley (IMPC) Recording 2024/02/09 Use cases for uPheno in the Alliance of Genome Resources and MGI Sue Bello (Alliance of Genome Resources, MGI) Recording"},{"location":"organization/outreach/#possible-topics","title":"Possible topics","text":"
Cross-species inference in Variant and Gene Prioritisation algorithms (Exomiser).
Cross-species comparison of phenotypic profiles (Monarch Initiative Knowledge Graph)
Cross-species data in biomedical knowledge graphs (Kids First)
"},{"location":"reference/core-concepts/","title":"Core concepts","text":""},{"location":"reference/core-concepts/#traits-and-phenotypes-the-conceptual-model","title":"Traits and phenotypes - the Conceptual model","text":""},{"location":"reference/core-concepts/#overview","title":"Overview","text":"
Here, we discuss the core concepts of the computational phenotype model underpinning the uPheno effort.
"},{"location":"reference/core-concepts/#table-of-contents","title":"Table of contents","text":"
\"Characteristics\" or \"qualities\" refer to an inherent or distinguishing characteristic or attribute of something or someone. It represents a feature that defines the nature of an object, organism, or entity and can be used to describe, compare, and categorize different things. Characteristics can be either qualitative (such as color, texture, or taste) or quantitative (such as height, weight, or age).
The Phenotype And Trait Ontology (PATO) is the reference ontology for general characteristics in the OBO world.
Some of the most widely use characteristics can be seen in the following tables
quality description example Length (PATO:0000122) A 1-D extent quality which is equal to the distance between two points. Mass (PATO:0000128) A physical quality that inheres in a bearer by virtue of the proportion of the bearer's amount of matter. Amount (PATO:0000070) The number of entities of a type that are part of the whole organism. Morphology (PATO:0000051) A quality of a single physical entity inhering in the bearer by virtue of the bearer's size or shape or structure.
Note from the authors: The descriptions above have been taken from PATO, but they are not very.. user friendly.
Characteristics such as the one above can be used to describe a variety of entities such as biological, environmental and social. We are specifically concerned with biological traits, which are characteristics that refer to an inherent characteristic of a biological entity, such as an organ (the heart), a process (cell division), a chemical entity (lysine) in the blood.
The Ontology of Biological Attributes (OBA) is the reference ontology for biological characteristics in the OBO world. There are a few other ontologies that describe biological traits, such as the Vertebrate Phenotype Ontology and the Ascomycete Phenotype Ontology (APO), but these are more species specific, and, more importantly, are not integrated in the wider EQ modelling framework.
Property Example term Definition Length OBA:VT0002544 The length of a digit. Mass OBA:VT0001259 The mass of a multicellular organism. Level OBA:2020005 The amount of lysine in blood. Morphology OBA:VT0005406 The size of a heart. "},{"location":"reference/core-concepts/#bearer-of-biological-characteristics","title":"Bearer of Biological Characteristics","text":"
In biological contexts, the term \"bearer\" refers to the entity that possesses or carries a particular characteristic or quality. The bearer can be any biological entity, such as an organism, an organ, a cell, or even a molecular structure, that exhibits a specific trait or feature. Some examples:
Organism as a Bearer:
Example: A specific tree (such as an oak tree) is the bearer of the characteristic 'height'.
Explanation: The tree as an organism carries or has the property of height, making it the bearer of this characteristic.
Organ as a Bearer:
Example: The heart of a mammal can be the bearer of the characteristic 'heart size'.
Explanation: Here, the heart is the organ that possesses the 'heart size' charactertistic. The characteristic ('heart size') is a quality of the heart itself.
Cell as a Bearer:
Example: A red blood cell is the bearer of the characteristic 'cell diameter'.
Explanation: The diameter is a property of the individual cell. Thus, each red blood cell is the bearer of its diameter measurement.
Molecular Structure as a Bearer:
Example: A DNA molecule can be the bearer of the characteristic 'sequence length'.
Explanation: The length of the DNA sequence is a property of the DNA molecule itself, making the molecule the bearer of this characteristic.
Genetic Trait as a Bearer:
Example: A fruit fly (Drosophila melanogaster) can be the bearer of a genetic trait like eye color.
Explanation: The organism (fruit fly) carries the genetic information that determines eye color, making it the bearer of this specific trait.
In each example, the \"bearer\" is the entity that has, carries, or exhibits a particular biological characteristic. This concept is fundamental in biology and bioinformatics for linking specific traits, qualities, or features to the entities that possess them, thereby enabling a clearer understanding and categorization of biological diversity and functions.
A phenotypic change refers to some deviation from reference morphology, physiology, or behavior. This is the most widely used, and most complicated category of phenotype terms for data specialists to understand.
Conceptually, a phenotypic change comprises:
a biological attribute (which includes a biological bearer)
an \"change\" modifier
(optionally) a directional modifier (increased / decreased)
a comparator
Biological attributes such as blood lysine amount (OBA:2020005) have been discussed earlier in this document. The most widely used change modifier used in practice is abnormal (PATO:0000460). This modifier signifies that the phenotypic change term describes a deviation that is abnormal, such as \"Hyperlysinemia\" (HP:0002161), which describes and increased concentration of lysine in the blood. Other modifiers include normal (PATO:0000461), which describes a change within in the normal range (sometimes interpreted as \"no change\"). A directional modifier like increased (PATO:0040043) or decreased (PATO:0040042). In practice, most of our \"characteristic\" terms have specialised directional variants such as decreased amount (PATO:0001997) which can be used to describe phenotypes.
"},{"location":"reference/core-concepts/#the-nuissance-of-implicit-comparators","title":"The nuissance of \"implicit comparators\"","text":"
Comparators are the most confusing aspects of phenotypic change. The first question someone has to ask when they see a concept describing is change like increased blood lysine levels is \"compared to what?\". Depending on biological context, the assumed comparators vary widely. For example, in clinical phenotyping, it is mostly assumed that a phenotypic feature corresponds to a deviation from the normal range, see HPO docs. However, it is just just as easily imaginable that HPO terms are used to describe change compared to a previous state of the same individual (increased tumor size compared to last time we checked). In research settings such as GWAS study annotations, HPO terms are used to annotate variants where a statistically significant change was observed compared to the general population. The same is true for many model phenotyping efforts such as MGI, where the situation is even further complicated that the comparator is not \"the general population\", but a control group. In summary, comparators can be:
The general population (\"wild type\" in research)
A non-representative sample of the general population (blood glucose values of all diabetes patients, a control group from the same mouse strain)
A previous state of a study subject (e.g. SNOMED).
And the compared charactertistics could be
Deviating from some notion of normality (abnormal)
A statistically significant change (including within normal)
No matter how much we want it - concepts describing phenotypic change will be used in many creative ways, and unfortunately, once the data hits your data analysis pipeline, you will likely not know for sure the nature of the comparator. Where you can, you should try to figure it out from the metata.
This sounds like bad news. However, keep one thing in mind: Phenotype associations (to anything, including genes) are rarely strictly causal. Even if a change is observed \"compared to some non-representative control\" there is likely to be some signal useful for downstream inference - somehow, the \"gene has something to do with the phenotype\".
"},{"location":"reference/core-concepts/#the-chaotic-terminology-around-phenotype","title":"The chaotic terminology around \"phenotype\"","text":"
In the clinical domain, many ontologies exist that define concepts that are very strongly related to our notion of \"phenotype\". In SNOMED, for example, \"clinical findings\" are defined as normal/abnormal observations, judgments, or assessments of patients (e.g. Abnormal urinalysis (finding)). For most analytic purposes, we think of SNOMEDs (and other medical terminologies) notion of clinical finding of something ortologous to our notion of \"phenotype\" (and their \"observale entity\" as a trait/biological attribute). However, if one gets into the weeds, many discrepencies in judgement can be observed, in particular when it comes to the separation from disease.
\"Phenotype\" is typically used in its \"singular\" form to describe the set of all observable characteristics of a subject. However, because we have over time gotten used to talking about \"cardiovascular phenotype\" and \"increased blood glucose level\", we have started using the plural form more, i.e. \"phenotypes\". We now tend to use the term \"phenotypic profile\" to describe the set of phenotypes that an organism exhibits at some point in time.
\"Phenotypic feature\" is a commonly used term that refers to the same idea, but mostly in the context of disease to describe an observable characteristic commonly associated with a disease.
\"Phenotypic abnormality\" is the formal term to describe a concept in the HPO, and is sometimes used to refer to the same idea in HPO-related papers. There is a bit of an assumption here, compared to the more general concepts described in this section, which is that the term should refer to a \"deviation from the normal range\", but, as described in the section of \"implicit comparators\", this assumption does not always hold in practice.
\"Phenotypic change\" is a recent invention by David Osumi-Sutherland in an attempt to subsume the ideas above, in particular to explicitly step back from the concept of \"deviation from normal\" to \"statistically significant deviation\" (which includes the normal range).
The Unified Phenotype Ontology (uPheno) is the reference ontology for biological abnormalities in the OBO world. There are a many species-specific ontologies in the OBO world, such as the Mammalian Phenotype Ontology (MP), the Human Phenotype Ontology (HPO) and the Drosophila Phenotype Ontology (DPO), see here.
Property Example term Definition Length UPHENO:0072215 Increased length of the digit. Mass UPHENO:0054299 Decreased multicellular organism mass. Level UPHENO:0034327 Decreased level of lysine in blood. Morphology UPHENO:0001471 Increased size of the heart. "},{"location":"reference/core-concepts/#diseases","title":"Diseases","text":"
Diseases are among the most important concepts in the phenotype data space. Phenotypes relate One big source of confusion in our community is the seperation of \"phenotypic features\" or changes from diseases. The HPO docs provide an explanation geared at clinicians to help them distinguish between the two. The quest on developing an operational definition is still ongoing, but for now, we recommend to go with the following basic assumptions:
There is a difference between disease and phenotype.
Phenotypes are features of diseases. Diseases can be associated with 1 or more phenotypic features. In the case of 1, we sometimes talk about \"isolated X\", for example \"Isolated Growth Hormone Deficiency (IGHD)\". IGHD is a condition where the pituitary gland produces insufficient growth hormone, leading to stunted growth.
Diseases, despite their grounding in biological reality, should be perceived mostly as social constructs that (adjusted from HPO docs):
Are used to capture a diagnosis (not just an observation - a judgement).
Are associated with a defined etiology (whether identified or as yet unknown, idiopathic). This is not about have some cause. This is about having a specific cause, even if it is unknown.
Have a defined time course (more or less well understood).
If treatments exist, there is a characteristic response to them. The key point is that the above are part of the disease definition.
In biological data curation, it\u2019s essential to differentiate between traits (observable characteristics such as \"blood glucose level\") and measurements (a process to observe such characteristics, e.g. \"blood glucose level assay\", \"BMI\"). Just from the term itself this is often difficult. \"Blood glucose level\" can refer both a measurement and a trait when taken out of context, but the ontologies they appear in should differenciate cleanly between the two. Here are some ways to distinguish them: - traits are - observable characteritics of an organism - can be qualitative (\"red eye colour\") or quantitative (\"35 cm tail length\") - measurements are - activties performed by an agent (such as a researcher) - involve the quantification or qualification of a specific trait - correspond to measurement instruments / techniques (such as assays, BMIs)
In practice, it is true that a lot of data records a wild mix of the two. It is the job of (semantic) data modeling specialists to clearly distinguish the two when integrating annotate data from sources with divergent curation practices.
"},{"location":"reference/core-concepts/#putting-it-all-together","title":"Putting it all together","text":"
Characteristics (A) and bearers of characteristics (B) are the core constituents of traits/biological attributes (C). Phenotypes are comprised of trait terms (C) combined with a modifier (D). Species-specific phenotypes (F), including phenotypic abnormalities defined in the Human Phenotype Ontology (HPO) are feature of diseases (G). Measurements (H), such as assays, quantify or qualify (measure) traits (C).
"},{"location":"reference/data-integration/","title":"Integrating phenotype data with the uPheno framework","text":""},{"location":"reference/data-integration/#integrating-phenotype-data-using-the-upheno-framework","title":"Integrating phenotype data using the uPheno framework","text":""},{"location":"reference/data-integration/#prerequisites","title":"Prerequisites","text":"
Before we get started, let's remind ourselves of the basic structure of phenotype data.
Characteristics (A) and bearers of characteristics (B) are the core constituents of traits/biological attributes (C). Phenotypes are comprised of trait terms (C) combined with a modifier (D). Species-specific phenotypes (F), including phenotypic abnormalities defined in the Human Phenotype Ontology (HPO) are feature of diseases (G). Measurements (H), such as assays, quantify or qualify (measure) traits (C).
Integrating all kinds of phenotype data into the \"uPheno framework\" is a complex process which we will break down in the following.
"},{"location":"reference/data-integration/#level-2-integration-knowledge","title":"Level 2 integration: Knowledge","text":""},{"location":"reference/data-integration/#important-relationships-wrt-to-phenotype-data","title":"Important relationships wrt to phenotype data","text":"
To avoid multiple duplicate classes for heart, lung, skin etc we map all classes to [Uberon] where this is applicable. For more divergent species such as fly and C elegans we use the appropriate species-specific ontology.
Currently there are a small number of highly specific classes in FMA that are being used and have no corresponding class in Uberon
We use the OWLAPI SyntacticLocalityModularityExtractor, via [OWLTools]. See the http://purl.obolibrary.org/obo/upheno/Makefile for details
"},{"location":"reference/phenotype-data/","title":"Phenotype data","text":""},{"location":"reference/phenotype-data/#phenotype-data-in-practice","title":"Phenotype Data in practice","text":""},{"location":"reference/phenotype-data/#overview","title":"Overview","text":"
The goals of this document are:
Give a sense of the contexts in which phenotype data is produced (research and clinical)
Give a sense of the shape of different styles of phenotype data
"},{"location":"reference/phenotype-data/#table-of-contents","title":"Table of contents","text":"
Some examples of phenotype data
Different shapes of phenotype data
Pre-coordinated
Post-coordinated
Standardised/non-standardized
Quantitative/qualitative
"},{"location":"reference/phenotype-data/#some-examples-of-phenotype-data","title":"Some examples of phenotype data","text":"Category Example datasets Example phenotype Gene to phenotype associations Online Mendelian Inheritance in Man (OMIM), Human Phenotype Ontology (HPO), Gene Ontology (GO) Achondroplasia (associated with FGFR3 gene mutations) Gene to disease associations The Cancer Genome Atlas (TCGA), Online Mendelian Inheritance in Man (OMIM), GWAS Catalog Breast invasive carcinoma (associated with BRCA1/BRCA2 mutations) Phenotype-phenotype semantic similarity Human Phenotype Ontology (HPO), Unified Medical Language System (UMLS), Disease Ontology (DO) Cardiac abnormalities (semantic similarity with congenital heart defects) Quantified trait data (QTL etc) NHGRI-EBI GWAS Catalog, Genotype-Tissue Expression (GTEx), The Human Protein Atlas Height (quantified trait associated with SNPs in genomic regions) Electronic health records Medical Information Mart for Intensive Care III (MIMIC-III), UK Biobank, IBM Watson Health Acute kidney injury (recorded diagnosis during ICU stay) Epidemiological datasets Framingham Heart Study, National Health and Nutrition Examination Survey (NHANES), Global Burden of Disease Study (GBD) Cardiovascular disease (epidemiological study of risk factors and disease incidence) Clinical trial datasets ClinicalTrials.gov, European Union Clinical Trials Register (EUCTR), International Clinical Trials Registry Platform (ICTRP) Treatment response (clinical trial data on efficacy and safety outcomes) Environmental exposure datasets Environmental Protection Agency Air Quality System (EPA AQS), Global Historical Climatology Network (GHCN), National Centers for Environmental Information Climate Data Online (NCEI CDO) Respiratory diseases (association with air pollutant exposure) Population surveys e.g., UK Biobank UK Biobank, National Health Interview Survey (NHIS), National Health and Nutrition Examination Survey (NHANES) Chronic diseases (population-based study on disease prevalence and risk factors) Behavioral observation datasets National Survey on Drug Use and Health (NSDUH), Add Health, British Cohort Study (BCS) Substance abuse disorders (survey data on drug consumption and addiction)"},{"location":"reference/phenotype-data/#different-shapes-of-phenotype-data","title":"Different shapes of phenotype data","text":"
Phenotype data comes in many different shapes and forms. In the following, we will describe some of the most common features of such data:
Pre-coordinated phenotype data is popular in the clinical domain, where a lot of observations are taken by a clinician and recorded as \"phenotypic abnormalities\" with the goal of eventual diagnosis.
Phenopackets such as the one below are an emerging standard to capture and sharing disease and phenotype information. Phenotypic features in particular are captured as so called \"pre-coordinated phenotype terms\" such as \"Attenuation of retinal blood vessels\" (HP:0007843). \"Pre-coordinated\" in this context means that the various aspects of the phenotype term, such as the bearer (\"retinal blood vessels\") and the characteristic (\"Attenuation\", or \"thinning/narrowing\"), and the modifier (in the case of HPO terms, simply abnormal), are combined (\"coordinated\") into a single term.
Phenopacket
{\n\"id\": \"PMID:23559858-Ajmal-2013-BBS1-IV-5/family_A\",\n\"subject\": {\n \"id\": \"IV-5/family A\",\n \"timeAtLastEncounter\": {\n \"age\": {\n \"iso8601duration\": \"P26Y\"\n }\n },\n \"sex\": \"MALE\",\n \"taxonomy\": {\n \"id\": \"NCBITaxon:9606\",\n \"label\": \"Homo sapiens\"\n }\n},\n\"phenotypicFeatures\": [\n {\n \"type\": {\n \"id\": \"HP:0007843\",\n \"label\": \"Attenuation of retinal blood vessels\"\n },\n \"evidence\": [\n {\n \"evidenceCode\": {\n \"id\": \"ECO:0000033\",\n \"label\": \"author statement supported by traceable reference\"\n },\n \"reference\": {\n \"id\": \"PMID:23559858\",\n \"description\": \"A family was reported in which two affected members had a splicing variant in BBS1, c.47+1G>T.\"\n }\n }\n ]\n },\n {\n \"type\": {\n \"id\": \"HP:0001513\",\n \"label\": \"Obesity\"\n },\n \"evidence\": [\n {\n \"evidenceCode\": {\n \"id\": \"ECO:0000033\",\n \"label\": \"author statement supported by traceable reference\"\n },\n \"reference\": {\n \"id\": \"PMID:23559858\",\n \"description\": \"A family was reported in which two affected members had a splicing variant in BBS1, c.47+1G>T.\"\n }\n }\n ]\n },\n {\n \"type\": {\n \"id\": \"HP:0000608\",\n \"label\": \"Macular degeneration\"\n },\n \"evidence\": [\n {\n \"evidenceCode\": {\n \"id\": \"ECO:0000033\",\n \"label\": \"author statement supported by traceable reference\"\n },\n \"reference\": {\n \"id\": \"PMID:23559858\",\n \"description\": \"A family was reported in which two affected members had a splicing variant in BBS1, c.47+1G>T.\"\n }\n }\n ]\n },\n {\n \"type\": {\n \"id\": \"HP:0000486\",\n \"label\": \"Strabismus\"\n },\n \"evidence\": [\n {\n \"evidenceCode\": {\n \"id\": \"ECO:0000033\",\n \"label\": \"author statement supported by traceable reference\"\n },\n \"reference\": {\n \"id\": \"PMID:23559858\",\n \"description\": \"A family was reported in which two affected members had a splicing variant in BBS1, c.47+1G>T.\"\n }\n }\n ]\n },\n {\n \"type\": {\n \"id\": \"HP:0001328\",\n \"label\": \"Specific learning disability\"\n },\n \"evidence\": [\n {\n \"evidenceCode\": {\n \"id\": \"ECO:0000033\",\n \"label\": \"author statement supported by traceable reference\"\n },\n \"reference\": {\n \"id\": \"PMID:23559858\",\n \"description\": \"A family was reported in which two affected members had a splicing variant in BBS1, c.47+1G>T.\"\n }\n }\n ]\n },\n {\n \"type\": {\n \"id\": \"HP:0000510\",\n \"label\": \"Rod-cone dystrophy\"\n },\n \"evidence\": [\n {\n \"evidenceCode\": {\n \"id\": \"ECO:0000033\",\n \"label\": \"author statement supported by traceable reference\"\n },\n \"reference\": {\n \"id\": \"PMID:23559858\",\n \"description\": \"A family was reported in which two affected members had a splicing variant in BBS1, c.47+1G>T.\"\n }\n }\n ]\n },\n {\n \"type\": {\n \"id\": \"HP:0001263\",\n \"label\": \"Global developmental delay\"\n },\n \"evidence\": [\n {\n \"evidenceCode\": {\n \"id\": \"ECO:0000033\",\n \"label\": \"author statement supported by traceable reference\"\n },\n \"reference\": {\n \"id\": \"PMID:23559858\",\n \"description\": \"A family was reported in which two affected members had a splicing variant in BBS1, c.47+1G>T.\"\n }\n }\n ]\n }\n],\n\"interpretations\": [\n {\n \"id\": \"PMID:23559858-Ajmal-2013-BBS1-IV-5/family_A\",\n \"progressStatus\": \"SOLVED\",\n \"diagnosis\": {\n \"disease\": {\n \"id\": \"OMIM:209900\",\n \"label\": \"BARDET-BIEDL SYNDROME 1; BBS1\"\n },\n \"genomicInterpretations\": [\n {\n \"subjectOrBiosampleId\": \"IV-5/family A\",\n \"interpretationStatus\": \"CAUSATIVE\",\n \"variantInterpretation\": {\n \"variationDescriptor\": {\n \"id\": \"clinvar:1324292\",\n \"geneContext\": {\n \"valueId\": \"ENSG00000174483\",\n \"symbol\": \"BBS1\",\n \"alternateIds\": [\n \"HGNC:966\",\n \"entrez:582\",\n \"ensembl:ENSG00000174483\",\n \"symbol:BBS1\"\n ]\n },\n \"vcfRecord\": {\n \"genomeAssembly\": \"GRCh37\",\n \"chrom\": \"11\",\n \"pos\": \"66278178\",\n \"ref\": \"G\",\n \"alt\": \"T\"\n },\n \"allelicState\": {\n \"id\": \"GENO:0000136\",\n \"label\": \"homozygous\"\n }\n }\n }\n }\n ]\n }\n }\n],\n\"metaData\": {\n \"created\": \"1970-01-01T00:00:00Z\",\n \"submittedBy\": \"HPO:probinson\",\n \"resources\": [\n {\n \"id\": \"hp\",\n \"name\": \"human phenotype ontology\",\n \"url\": \"http://purl.obolibrary.org/obo/hp.owl\",\n \"version\": \"2018-03-08\",\n \"namespacePrefix\": \"HP\",\n \"iriPrefix\": \"http://purl.obolibrary.org/obo/HP_\"\n },\n {\n \"id\": \"pato\",\n \"name\": \"Phenotype And Trait Ontology\",\n \"url\": \"http://purl.obolibrary.org/obo/pato.owl\",\n \"version\": \"2018-03-28\",\n \"namespacePrefix\": \"PATO\",\n \"iriPrefix\": \"http://purl.obolibrary.org/obo/PATO_\"\n },\n {\n \"id\": \"geno\",\n \"name\": \"Genotype Ontology\",\n \"url\": \"http://purl.obolibrary.org/obo/geno.owl\",\n \"version\": \"19-03-2018\",\n \"namespacePrefix\": \"GENO\",\n \"iriPrefix\": \"http://purl.obolibrary.org/obo/GENO_\"\n },\n {\n \"id\": \"ncbitaxon\",\n \"name\": \"NCBI organismal classification\",\n \"url\": \"http://purl.obolibrary.org/obo/ncbitaxon.owl\",\n \"version\": \"2018-03-02\",\n \"namespacePrefix\": \"NCBITaxon\",\n \"iriPrefix\": \"http://purl.obolibrary.org/obo/NCBITaxon_\"\n },\n {\n \"id\": \"eco\",\n \"name\": \"Evidence and Conclusion Ontology\",\n \"url\": \"http://purl.obolibrary.org/obo/eco.owl\",\n \"version\": \"2018-11-10\",\n \"namespacePrefix\": \"ECO\",\n \"iriPrefix\": \"http://purl.obolibrary.org/obo/ECO_\"\n },\n {\n \"id\": \"omim\",\n \"name\": \"Online Mendelian Inheritance in Man\",\n \"url\": \"https://www.omim.org\",\n \"version\": \"2018-03-08\",\n \"namespacePrefix\": \"OMIM\",\n \"iriPrefix\": \"https://omim.org/entry/\"\n },\n {\n \"id\": \"clinvar\",\n \"name\": \"Clinical Variation\",\n \"url\": \"https://www.ncbi.nlm.nih.gov/clinvar/\",\n \"version\": \"2023-04-06\",\n \"namespacePrefix\": \"clinvar\",\n \"iriPrefix\": \"https://www.ncbi.nlm.nih.gov/clinvar/variation/\"\n }\n ],\n \"phenopacketSchemaVersion\": \"2.0.0\"\n}\n}\n
Apart from clinical diagnostics, pre-coordinated phenotype terms are used in many other contexts such as model organism research (e.g. IMPC) or the curation of Genome Wide Association Studies.
Post-coordinated phenotype curation simply means that the different constituents of phenotype (characteristic, bearer, modifier etc) are captured individually. This has certain advantages. For example, the phenotype space is enormous, as you can measure variations in many observable charactertics from chemical entities present in the blood, the microbiome to a host of morphological and developmental abnormalities. Instead of having individual (controlled vocabulary) terms for increased level of X, decreased level X, abnormal level of X, increased level of X in blood for thousands of chemical compounds synthesized by the human body, you just have \"increased level\", \"blood\" and all the chemical compounds.
There are at least three flavours of post-coordinated phenotype curation prevalent in the biomedical domain, four if you count quantified phenotypes:
Trait + modifier
Bearer only
Characteristics + modifier + bearer
Trait + modifier pattern is used for example by databases such as the Saccharomyces Genome Database (SGD). Here are some examples:
APO:0000002 (abnormal) and APO:0000003 (decreased) are modifiers.
APO:0000087 (resistance to chemicals), APO:0000224 (RNA accumulation), APO:0000052 (cell size) are biological attributes/traits.
CHEBI:78661 is recorded as an experimental condition, but should probably be interpreted as part of the bearer expression.
Note: SGD has different kinds of phenotype data, and it should be carefully evaluated which one it is.
Data was obtained from the Alliance of Genome Resources on the 30.03.2023 and simplified for illustration.
The bearer-only pattern is used by many databases, such as Flybase. In the data, we only find references of bearers, such as anatomical entities or biological processes. Instead of explicitly stating phenotypic modifiers (abnormal, morphology, changed), it is implicit in the definition of the dataset.
FBbt:00000184 (embryonic telson) and FBbt:00001727 (larva) are bearer terms.
The modifier is implicit in the data rather than explicitly stated. For example, Flybase states on their website about the Dmel\\torrv66 Allele (FBal0016988) that the \"phenotype manifests in the embryonic telson\".
Note: FlyBase has different kinds of phenotype data (including pre-coordinated), and it should be carefully evaluated which one is which prior to integration.
Data was obtained from the Alliance of Genome Resources on the 30.03.2023 and simplified for illustration.
The most complex pattern for phenotype descriptions which essentially decomposes the entire phenotype expression into atomic consituents can be found, for example, in the The Zebrafish Information Network (ZFIN).
Examples:
Fish ID Affected Structure or Process 1 subterm ID Affected Structure or Process 1 subterm Name Post-composed Relationship ID Post-composed Relationship Name Affected Structure or Process 1 superterm ID Affected Structure or Process 1 superterm Name Phenotype Keyword ID Phenotype Keyword Name Phenotype Tag Affected Structure or Process 2 subterm ID Affected Structure or Process 2 subterm name Post-composed Relationship (rel) ID Post-composed Relationship (rel) Name Affected Structure or Process 2 superterm ID Affected Structure or Process 2 superterm name Publication ID ZDB-FISH-150901-29105 ZFA:0009366 hair cell BFO:0000050 part_of ZFA:0000051 otic vesicle PATO:0000374 increased distance abnormal ZFA:0009366 hair cell BFO:0000050 part_of ZFA:0000051 otic vesicle ZDB-PUB-171025-12 ZDB-FISH-150901-29105 ZFA:0009366 hair cell BFO:0000050 part_of ZFA:0000051 otic vesicle PATO:0000374 increased distance abnormal ZFA:0009366 hair cell BFO:0000050 part_of ZFA:0000051 otic vesicle ZDB-PUB-171025-12 ZDB-FISH-150901-11537 ZFA:0000051 otic vesicle PATO:0001905 has normal numbers of parts of type normal ZFA:0009366 hair cell BFO:0000050 part_of ZFA:0000051 otic vesicle ZDB-PUB-150318-1 ZDB-FISH-150901-18770 ZFA:0000119 retinal inner nuclear layer PATO:0002001 has fewer parts of type abnormal ZFA:0009315 horizontal cell BFO:0000050 part_of ZFA:0000119 retinal inner nuclear layer ZDB-PUB-130222-28 ZDB-FISH-190806-7 BSPO:0000084 ventral region BFO:0000050 part_of ZFA:0000101 diencephalon PATO:0002001 has fewer parts of type abnormal ZFA:0009301 dopaminergic neuron BFO:0000050 part_of ZFA:0000101 diencephalon ZDB-PUB-190216-5 ZDB-FISH-190807-7 BSPO:0000084 ventral region BFO:0000050 part_of ZFA:0000101 diencephalon PATO:0001905 has normal numbers of parts of type normal ZFA:0009301 dopaminergic neuron BFO:0000050 part_of ZFA:0000101 diencephalon ZDB-PUB-190216-5 ZDB-FISH-190807-8 BSPO:0000084 ventral region BFO:0000050 part_of ZFA:0000101 diencephalon PATO:0002001 has fewer parts of type abnormal ZFA:0009301 dopaminergic neuron BFO:0000050 part_of ZFA:0000101 diencephalon ZDB-PUB-190216-5 ZDB-FISH-150901-29105 ZFA:0000101 diencephalon PATO:0001555 has number of normal ZFA:0009301 dopaminergic neuron BFO:0000050 part_of ZFA:0000101 diencephalon ZDB-PUB-161120-7 ZDB-FISH-210421-9 ZFA:0009290 glutamatergic neuron BFO:0000050 part_of ZFA:0000008 brain PATO:0040043 increased proportionality to abnormal ZFA:0009276 GABAergic neuron BFO:0000050 part_of ZFA:0000008 brain ZDB-PUB-191011-2 ZDB-FISH-210421-9 ZFA:0009290 glutamatergic neuron BFO:0000050 part_of ZFA:0000008 brain PATO:0040043 increased proportionality to abnormal ZFA:0009276 GABAergic neuron BFO:0000050 part_of ZFA:0000008 brain ZDB-PUB-191011-2
Lets break down the second to last row:
ZFA:0009290 (glutamatergic neuron): The primary entity whose characteristic is being observed
BFO:0000050 (part of): a relation used to connect the hair cell to the structure its part of
ZFA:0000008 (brain): the location of the primary entity being observed
PATO:0040043 (increased proportionality to): the modified characteristic being observed.
abnormal: the change modifier (note: not an ontology term)
ZFA:0009276 (GABAergic neuron): the secondary entity being observed in relation to which the characteristic is measured
ZFA:0000008 (brain): the location of the secondary entity
The interested reader may look at an integrated version of that huge post-coordinated expression here (brain increased proportionality to glutamatergic neuron GABAergic neuron brain, abnormal - ZP:0141834).
Data was obtained from ZFIN (Phenotype of Zebrafish Genes) on the 30.03.2023 and simplified for illustration.
As one can see in the last example, bearers can be anything from simple atomic entities to arbitrarily complex compositions:
\"lysine\" (lysine)
\"lysine in the blood\" (lysine part_of blood)
\"lysine in heart muscle cells\" (lysine part_of cell part_of (muscle part of heart))
\"lysine in the cytoplasm of heart muscle cells\" (lysine part_of (cytoplasm part_of (cell part_of (muscle part of heart))))
Phenotype data can be standardised to varying degrees. It is not uncommon for data to be completely unstandardised. Unfortunately, only a fraction of the available data is actually annotated using terms from controlled phenotype ontologies. Here are some of the more \"typical\" kinds of data on the standardised/non-standardised spectrum:
Free text in clinical notes and scientific publications
Free text in specific database fields (for example a \"height\" column in a table about measurements of Giraffes)
Controlled but non-standardised vocabulary like enums in a datamodel (for example the keyword \"abnormal\" in the ZFIN example above)
Controlled standardised vocabulary (like all the examples on this page)
Ontology terms (controlled vocabulary terms with well defined semantics - all the examples on this page)
Qualitative and quantitative phenotype data represent two fundamental ways of describing characteristics or traits in biology, each providing different types of information:
Qualitative Phenotype Data:
Nature: This type of data describes qualities or characteristics that are observed but not measured with numbers. It often involves categorical or descriptive information.
Examples: The presence or absence of a specific physical trait (like eye color or wing shape in animals) or types of behavior (aggressive vs. passive).
Analysis: Qualitative data is analyzed by categorization and identification of patterns or variations. It is more about the 'type' or 'kind' of trait rather than its 'amount'.
Interpretation: Since it's descriptive, this data relies on subjective interpretation and classification.
Quantitative Phenotype Data:
Nature: This data is numerical and quantifies traits. It involves measurements of characteristics, often allowing for more precise and objective analysis.
Examples: Height, weight, blood pressure, cholesterol levels, or the number of fruit produced by a plant. Quantitative traits can often be measured on a continuous scale, for example height of 35 cm, weight of 67 KG or blood pressure of 120/80.
Analysis: It involves statistical analysis, such as calculating mean, median, standard deviation, and applying various statistical tests. It allows for a more objective and replicable assessment.
Interpretation: Quantitative data provides a more concrete and measurable understanding of traits, making comparisons and statistical testing more straightforward.
Qualitative data is descriptive and categorical, while quantitative data is numerical and measurable. Both types are essential for a comprehensive understanding of phenotypic traits, each offering unique insights into biological variation and complexity.
"},{"location":"reference/phenotype-ontology-alignment/","title":"Aliging species specific phenotype ontologies","text":"
Phenotype ontologies use different reference ontologies for their EQs. Everything in uPheno is integrated towards a common set of reference ontologies, in particular Uberon and CL. In order to integrate species-independent anatomy ontologies we employ the following workflow for phenotype ontologies:
Create a base-plus module from the ontology
Rename all Uberon-aligned entities using ROBOT rename. This replaces basically species specific anatomy references with Uberon anatomy references
Delete all species specific references from uPheno (FBBT, XAO, ZFA, etc). This also deletes all EQs which have non-Uberon references.
For all remaining species-specific anatomy terms, we retain only the link to the nearest Uberon term.
"},{"location":"reference/phenotype-ontology-alignment/#rules-for-phenotype-ontologies-to-be-integrated","title":"Rules for phenotype ontologies to be integrated","text":"
Every phenotype ontology must export a base module at the proper PURL location
Every phenotype ontology must export a upheno export module at the proper PURL location
When two classes are merged in uPheno based on a cross-species mapping, we assert the most general common ancestor as parent.
This includes a phenotype class for every anatomy class - the IRI is suffixed with \"PHENOTYPE\". Using these ontologies, Uberon and CL phenotypes make the groupings.
The structure roughly follows that of the [MP]. The worm anatomy is used.
"},{"location":"reference/components/wbphenotype/#editing-the-axioms","title":"Editing the axioms","text":"
Currently the source is wbphenotype/wbphenotype-equivalence-axioms.obo, the OWL is generated from here. We are considering switching this around, so the OWL is edited, using Protege.
The ZP differs considerably from [HP], [MP] and others. ZFIN do not annotate with a pre-composed phenotype ontology - all annotations compose phenotypes on-the-fly using a combination of PATO, ZFA, GO and other ontologies.
We use these combinations to construct ZP on the fly, by naming each distinct combination, assigning it an ID, and placing it in the hierarchy.
The process is described here:
Sebastian K\u00f6hler, Sandra C Doelken, Barbara J Ruef, Sebastian Bauer, Nicole Washington, Monte Westerfield, George Gkoutos, Paul Schofield, Damian Smedley, Suzanna E Lewis, Peter N Robinson, Christopher J Mungall (2013) Construction and accessibility of a cross-species phenotype ontology along with gene annotations for biomedical research F1000Research
The OWL formalism for ZFIN annotations is described here:
[https://docs.google.com/document/d/1Vbokc9aFHR4awNE6DrrLtgpE6axeTS4VEfxqDHsWyPQ/edit# Mapping ZFIN phenotypes to OWL]
Because the latter class definition includes qualifier some abnormal, the SubClassOf axiom will not be entailed unless the qualifier is explicitly stated or inferred
Much has been written on the subject of representing absence. Before diving into the logical issues it is worth examining patterns in existing phenotype ontologies to understand what user expectations may typically be for absence.
"},{"location":"reference/modelling/absence/#details","title":"Details","text":""},{"location":"reference/modelling/absence/#strict-logical-absence-vs-absence-of-some","title":"Strict logical absence vs absence of some","text":"
It is not uncommon to see patterns such as
From a strict logical perspective, this is inverted. \"absent incisors\" surely means \"absence of all incisors\", or put another way \"the animal has no incisors\". Yet it would be possible to have an animal with *absent* lower incisors and *present* upper incisors, yielding what seems a contradiction (because the subClass axiom would say this partial-incisor animal lacked all incisors).
If the ontology were in fact truly modeling \"absence of *all* S\" then it would lead to a curious ontology structure, with the typical tree structure of the anatomy ontology representing S inverted into a polyhierarchical fan in the absent-S ontology.
From this it can be cautiously inferred that the intent of the phenotype ontology curator and user is in fact to model \"absence of *some* S\" rather than \"absence of *all* S\". This is not necessarily a universal rule, and the intent may vary depending on whether we are talking about a serially repeated structure or one that typically occurs in isolation. The intent may also be to communicate that a *significant number* of S is missing.
"},{"location":"reference/modelling/absence/#absence-as-a-type-of-morphology","title":"Absence as a type of morphology","text":"
It is also not uncommon to see patterns such as:
Again, from a strict logical perspective this is false. If the spleen is absent then what does the \"morphology\" of the parent refer to?
However, this inference is clearly a desirable one from the point of view of the phenotype ontology editors and users, as it is common in ontologies for a variety of structures. For example:
And:
These patterns can be formally defended on developmental biology grounds. \"absence\" here is _not_ equivalent to logical absence. It refers specifically to developmental absence.
Furthermore, strict logical absence leads to undesirable inferences. It would be odd to include a nematode worm as having the phenotype \"spleen absent\", because worms have not evolved spleens. But the logical description of not having a spleen as part fets a worm.
Similarly, if the strict cardinality interpretation were intended, we would expect to see:
i.e. if you're missing your entire hindlegs, you're *necessarily* missing your femurs. But it must be emphatisized that this is *not* how phenotype ontologies are classified. This goes for a wide range of structures and other relationship types. In MP, \"absent limb buds\" are *not* classified under \"absent limbs\", even though it is impossible for a mammal to have limbs without having had limb buds.
"},{"location":"reference/modelling/absence/#absence-as-part-of-a-size-morphology-spectrum","title":"Absence as part of a size-morphology spectrum","text":"
The existing treatment of absence can be formally defended morphologically by conceiving of a morphological value space, with \"large\" at one end and \"small\" at the other. As we get continuously smaller, there may come an arbitrary point whereby we say \"surely this is no longer a limb\" (and of course, we are not talking about a pure geometrical size transformation here - as a limb reaches extreme edges of a size range various other morphological changes necessarily happen). But this cutoff is arguably arbitrary, and the resulting discontinuity causes problems. It is simpler to treat absence as being one end of a size scale.
This is barely touching the subject, and is intended to illustrate that things may be more subtle than naively treating words like \"absent\" as precisely equivalent to cardinality=0. An understanding of the medical, developmental and evolutionary contexts are absolutely required, together with an understanding of the entailments of different logical formulations.
Even though existing phenotype ontologies may not be conceived of formally, it is implicit than they do not model absence as being equivalent to cardinality=0 / not(has_part), because the structure of these ontologies would look radically different.
Link to Jim Balhoff's PhenoDay paper and discussion
Here's the link: http://phenoday2014.bio-lark.org/pdf/11.pdf
"},{"location":"reference/qc/odk_checks/","title":"ODK: Basic Quality Control","text":""},{"location":"tutorials/analysis/","title":"Using uPheno in Data Analysis","text":""},{"location":"tutorials/analysis/#using-oba-and-upheno-for-data-analysis","title":"Using OBA and uPheno for data analysis","text":"
Familiarise yourself with the basics of phenotype integration
Familiarise yourself with the use cases
"},{"location":"tutorials/curation/","title":"Using uPheno in Curation","text":""},{"location":"tutorials/curation/#using-oba-and-upheno-in-data-curation","title":"Using OBA and uPheno in data curation","text":"

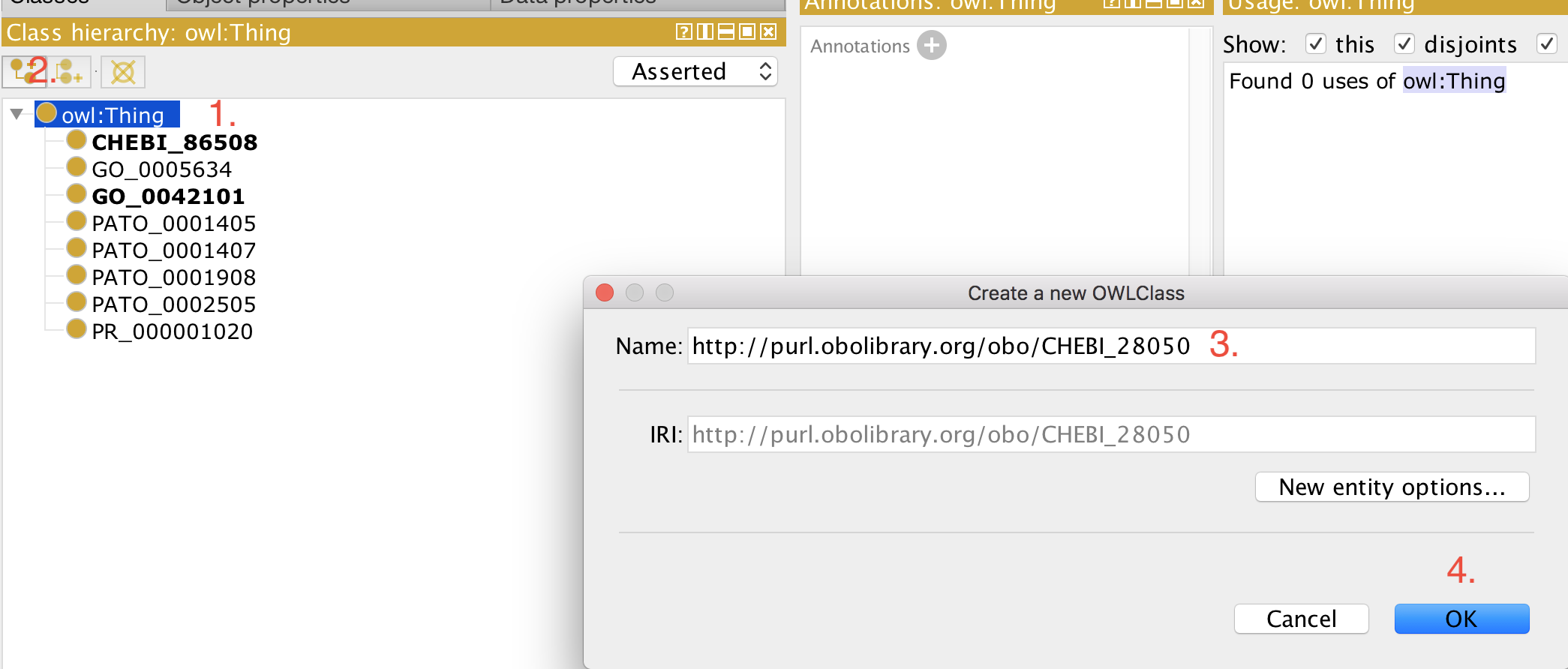

{kind=link}