-
Notifications
You must be signed in to change notification settings - Fork 0
Commit
This commit does not belong to any branch on this repository, and may belong to a fork outside of the repository.
- Loading branch information
Showing
5 changed files
with
242 additions
and
118 deletions.
There are no files selected for viewing
This file contains bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
This file contains bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
This file contains bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
| Original file line number | Diff line number | Diff line change |
|---|---|---|
| @@ -1,115 +1,34 @@ | ||
| >  | ||
| # biotools | ||
|
|
||
| Tools designed to perform and work with cluster analysis (including Tocher's algorithm), as well as some useful miscellaneous tools for evaluating clustering outcomes, such as a specific coefficient of cophenetic correlation, discriminant analysis, the Box'M test and the Mantel's permutation test. A new approach for calculating the power of Mantel's test is implemented. biotools also contains the new tests for genetic covariance components. An approach for predicting spatial gene diversity is implemented. Some of the main tools are described in the next sections. | ||
|
|
||
| Target audience: agronomists, biologists and researchers of related fields. | ||
|
|
||
| ## Importance of variables: Singh's criterion | ||
|
|
||
| The function singh() runs the method proposed by Singh (1981) for determining the importance of variables based on the squared generalized Mahalanobis distance. In his approach, the importance of the j-th variable (j = 1, 2, ..., p) on the calculation of the distance matrix can be obtained by: | ||
|
|
||
|  | ||
|
|
||
| \noindent where 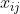 and  are the observation taken at the i-th and i'-th objects (individuals) for the j-th variable,  is the p-variate vector of the i-th object and  is the j-th column of the inverse of the covariance matrix. | ||
|
|
||
| The output of singh() is basically a matrix containing every  and its relative value. | ||
|
|
||
| ## Tocher's algorithm | ||
|
|
||
| biotools contains the method suggested by K.D. Tocher (Rao 1952) for clustering objects, which consists of an optimization method that follows the algorithm: | ||
|
|
||
| 1. (Input) Take a distance matrix, let us say , of size n. | ||
| 2. Define a clustering criterion, i.e., a limit of distance at which to evaluate the inclusion of objects in a cluster in formation. Usually, this criterion is defined as follows:  be a vector of size n containing the lowest distances involving each object, then take  to be the clustering criterion. | ||
| 3. Locate the pair of objects with the lowest distance in  to be the starting cluster. | ||
| 4. Compute the average distance  of the starting cluster, say k, to a certain object, say j, that shows the lowest pairwise distance with each object in the starting cluster. | ||
| 5. Compare the statistic evaluated at the step 4 with 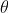. Then, add the new object to the starting cluster if . Otherwise, go to step 3 not considering the objects already clustered and start a new cluster. | ||
|
|
||
| The process continues until the last remaining object is evaluated and either included in the last cluster formed or allocated to its own separated cluster. The function tocher() performs optimization clustering and returns an object of class 'tocher', which contains the clusters formed, a numeric vector indicating the cluster of each object, a matrix of cluster distances and also the input, a 'dist' object. | ||
|
|
||
| ## Cluster distances | ||
|
|
||
| After obtaining the clusters, it might be useful to know how divergent they are from each other. In this context, cluster distances are calculated from the original distance matrix through the function distClust(). An intracluster distance is calculated by averaging all pairwise distances among objects in the cluster concerned. Likewise, the distance between two clusters is calculated by averaging all pairwise distances among objects in these clusters. | ||
|
|
||
| ## Cophenetic correlation | ||
|
|
||
| Clustering validation is widely made for hierarchical and iterative methods. Some measures of internal validation for a Tocher's clustering outcome currently implemented on biotools. | ||
|
|
||
| The approach presented by Silva (2013) is implemented by taking the cluster distances in order to build a cophenetic matrix for clustering performed through the Tocher's method. Their approach consists of taking the cophenetic distance among objects located in the same cluster as the intracluster distance and the cophenetic distance between objects of different clusters as the intercluster distance. Then, the Pearson's correlation between the elements of the original and cophenetic matrix can be taken as a cophenetic correlation. The function to be called is cophenetic(), whose input is an object of class 'tocher' and its output an object of class 'dist'. | ||
|
|
||
| ## Box's M-test | ||
|
|
||
| When clusters are formed, one might be interested in verifying if covariance matrices of the clusters can be considered homogeneous, especially if one intends to perform a linear discriminant analysis using a pooled matrix. In this case, the well known Box's M-test can be applied. The function boxM() performs the test using an approximation of the  distribution, where , and k is the number of covariance matrices. Users should be aware that all clusters must have a positive definite covariance matrix. | ||
|
|
||
| ## Discriminant analysis based on Mahalanobis distance | ||
|
|
||
| A simple and efficient object classification rule is based on Mahalanobis distance. It consists of calculating the squared generalized Mahalanobis distances of each multivariate observation to the centre of each cluster. biotools performs this sort of discriminant analysis through D2.disc(), as follows: consider  as the Mahalanobis distance from the i-th (i = 1, 2, ..., n) observation in the j-th (j = 1, 2, ..., k) cluster to the probable j'-th cluster. Now consider 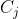 the random variable that represents the cluster at which this observation lies. The predicted class  for this concerned observation is the one such that . | ||
|
|
||
| D2.disc() outputs the Mahalanobis distances from each observation to the centre of each cluster, the pooled covariance matrix for clusters and the confusion matrix, which contains the number of correct classifications in the diagonal cells and misclassifications off-diagonal. In addition, a column misclass indicates (with an asterisk) where there was disagreement between the original classification (grouping) and the predicted one (pred). | ||
|
|
||
| # Installation | ||
|
|
||
| You can install and load the released version of biotools from GitHub with: | ||
|
|
||
| ```r | ||
| devtools::install_github("arsilva87/biotools") | ||
|
|
||
| library(biotools) | ||
| ``` | ||
|
|
||
| # Example of Tocher's clustering | ||
|
|
||
| Seventeen garlic genotypes from a Germoplasm Bank were evaluated in terms of agronomic variables in order to obtain contasting clusters and to eliminate replicates. A matrix containing the Generalized Mahalanobis distances among genotypes is given in 'garlicdist'. Tocher's method is applied to determine clusters. | ||
|
|
||
| ```r | ||
| data(garlicdist) | ||
|
|
||
| (toc <- tocher(garlicdist)) | ||
|
|
||
| Tocher's Clustering | ||
| Call: tocher.dist(d = garlicdist) | ||
| Cluster algorithm: original | ||
| Number of objects: 17 | ||
| Number of clusters: 6 | ||
| Most contrasting clusters: cluster 3 and cluster 5, with | ||
| average intercluster distance: 11.78786 | ||
| $`cluster 1` | ||
| [1] 8 9 12 4 10 2 7 15 | ||
| $`cluster 2` | ||
| [1] 1 6 14 | ||
| $`cluster 3` | ||
| [1] 11 13 | ||
| $`cluster 4` | ||
| [1] 3 5 | ||
| $`cluster 5` | ||
| [1] 16 | ||
| $`cluster 6` | ||
| [1] 17 | ||
| toc$distClust # cluster distances | ||
| cluster 1 cluster 2 cluster 3 cluster 4 cluster 5 cluster 6 | ||
| cluster 1 1.745434 4.333530 3.264753 7.070493 8.816863 3.045773 | ||
| cluster 2 4.333530 1.930265 7.525301 4.156222 3.476651 3.560654 | ||
| cluster 3 3.264753 7.525301 2.317785 8.019206 11.787861 6.596850 | ||
| cluster 4 7.070493 4.156222 8.019206 2.324152 4.043741 8.484307 | ||
| cluster 5 8.816863 3.476651 11.787861 4.043741 0.000000 5.441962 | ||
| cluster 6 3.045773 3.560654 6.596850 8.484307 5.441962 0.000000 | ||
| cof <- cophenetic(toc) # cophenetic matrix | ||
| cor(cof, garlicdist) # cophenetic correlation coefficient | ||
| [1] 0.9086886 | ||
| ``` | ||
| # Contributions and bug reports | ||
| **biotools** is an ongoing project. Then, contributions are very welcome. If you have a question or have found a bug, please open an  or reach out directly by e-mail: anderson.silva@ifgoiano.edu.br. | ||
| <!-- badges: start --> | ||
| [](https://CRAN.R-project.org/package=biotools) | ||
|  | ||
| <!-- badges: end --> | ||
|
|
||
|  | ||
|
|
||
| The package **biotools** has been developed for helping biologists, ecologists and researchers of related fields to perform optimization cluster analysis, specifically using Tocher's methods, as well as to evaluate the quality of the clustering. For this last purpose, some new and standard methodologies are supplied: the cophenetic correlation coefficient, Box's M-test for equality of covariance matrices and discriminant analysis based on Mahalanobis distance. These two latter can also be used upon clustering obtained with other methods, including the hierarchical ones. | ||
|
|
||
| Users can also find some other useful tools, such as a function for performing the well known Mantel's test and for computing its *simulated power*, a function that allows one to perform path analysis dealing with collinearity and a function for determining the minimum sample size in a very general way. | ||
|
|
||
| # Getting started | ||
|
|
||
| You can install the released version on CRAN directly from the R console, with: | ||
|
|
||
| ```r | ||
| install.packages("biotools") | ||
| ``` | ||
| Or you can install the development version from GitHub, using: | ||
|
|
||
| ```r | ||
| devtools::install_github("arsilva87/biotools") | ||
| ``` | ||
| Afterwards, just load it and it will be ready to use. | ||
|
|
||
| ```r | ||
| library("biotools") | ||
| ``` | ||
|
|
||
| Check the "Articles" menu to see illustrations of some of its functions. In the "Reference" menu you can find the complete documentation and examples of all functions and data sets. | ||
|
|
||
| # Contact | ||
|
|
||
| **biotools** is an ongoing project. Contributions are very welcome. If you have a question or have found a bug, please open an  or reach out directly by e-mail: anderson.silva@ifgoiano.edu.br. |
This file contains bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
| Original file line number | Diff line number | Diff line change |
|---|---|---|
| @@ -0,0 +1,3 @@ | ||
| template: | ||
| params: | ||
| bootswatch: simplex |
Oops, something went wrong.