TBA is a multi-functional machine learning tool for identifying transcription factors associated with genomic features. Specifically, TBA can be applied to:
- ChIP-seq targeting a transcription to identify collaborative binding partners for a given transcription factor.
- DNAse-seq and ATAC-seq to identify transcription factors associated with open chromatin
- GRO-seq and other assays measuring enhancer activity to identify transcription factors associated with enhancer activty
- Predict the effect of genetic variation in any of the above contexts.
TBA takes a set of loci that are of interest as input. First, the genomic sequence of each loci of interest are retrieved. Next, TBA selects a set of GC-matched background loci. For each locus of interest and background locus, TBA calculates the best match to hundreds of DNA binding motifs, and quantifies the quality of the match as the motif score (aka log likelihood ratio score). To allow for degenerate motifs, all motif matches scoring over zero are considered. The motif scores are then used to train the TBA model to distinguish loci of interest from background loci. TBA scores the probability of observing binding at a sequence by computing a weighted sum over all the motif scores for that sequence. The weight for each motif is learned by iteratively modifying the weights until the model’s ability to differentiate binding sites from background loci no longer improves. The final motif weight measures whether the presence of a motif is correlated with TF binding.
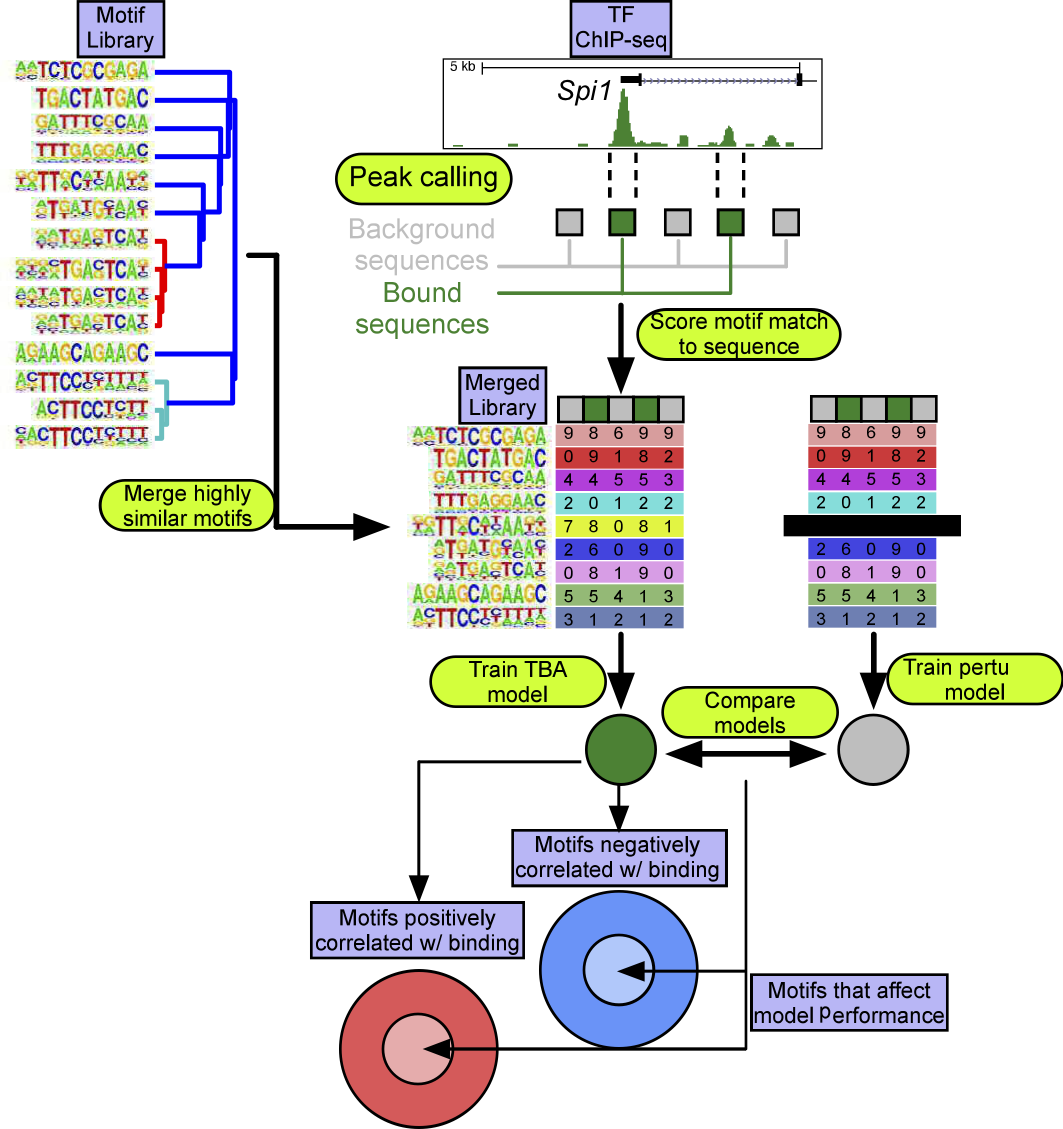
TBA uses a programatically curated library of motifs to reduce the effects of multiple collinearity, which can be problematic for machine learning models. You can view and download the motifs at: homer.ucsd.edu/jtao/merged_motifs/allList.html](http://homer.ucsd.edu/jtao/merged_motifs/allList.html "Motif Library")
TBA can be used on most computers running a Unix operating system (eg. macOS and Ubuntu). TBA models can be trained on a data set containing ~30k genomic loci in a reasonable time frame (~2 hours) on a modern laptop (8 GB DDR3 RAM, 2.5 GHz CPU).
Download the latest release of TBA (source code) and decompress the download. Next add the "model_training" directory to your PATH environment variable. You can use the nano command to edit your .bashrc or .bash_profile file to modify how your PATH variable is set. You should add a line in your .bashrc or .bash_profile file to say
PATH=$PATH:/path/to/tba/model_training; export PATH
TBA depends on several publicly available software packages as well as data resources. Please install the following software packages:
- Required:
- Python 3.5
- sklearn 0.19.0
- scipy
- pandas 0.20.3
- pandas 1.14.0
- biopython 0.17
- joblib 0.11
- Required packages bundled with most Python installations
- multiprocessing
- os
- time
- inspect
- argparse
- sys
- pickle
- Recommended:
- Homer - for processing most types of genomic data
- homer.ucsd.edu
- Please see our manuscript for detail
- IDR - for identifying highly reproducible peaks in ChIP-seq and ATAC-seq data
- https://github.com/nboley/idr
- Please see our manuscript for detail
- Seaborn - for data visualization
- Homer - for processing most types of genomic data
TBA requires the genomic sequence of loci of interest. You can download the the genomic sequence of most organisms with a sequenced genome from the UCSC Genome Browser Gateway. Create a directory at where you installed TBA with the name of the genome (eg: /path/to/tba/download/model/training/hg38 for the hg38 build of the human genome); within this new directory, download the fasta file for each chromosome separately. The file structure should look like this:
/path/to/tba/model_training/hg38/
chr1.fa
chr2.fa
chr3.fa
...
chrY.fa
If you have your own preferred way of retrieving genomic sequence, TBA can use a FASTA file containing the sequences of regions of interest instead of a BED file. However, TBA needs a genome to be installed using the directions above to select background sequences. Supposing you have your preferred way of generating a set of background sequences, TBA can use custom background sequences in FASTA format.
TBA is accessible as a series of command line (aka terminal) scripts. The easiest way to train a TBA model is to use the train_model_default.sh command. train_model_default.sh will run all TBA commands necessary to train a TBA with default parameters. You can invoke the command like this:
train_model_default.sh mouse_pu1_peaks.bed mm10 path_to_output The script will create a script at path_to_output/run.sh and execute it. run.sh will have correctly formatted TBA commands for each step. You can modify this script file with custom parameters if needed. Output files will be created at path_to_output. The run.sh script will look something like this:
# Extract the genomic coordinates of your regions of interest.
# You can skip this step if you already have a fasta file.
# Run time: several minutes
extract_sequences.py /path/to/bed_file.bed genome /path/to/output/fasta_file.fasta
# Generate GC content matched background coordinates.
# You can skip this step if you already have your background coordinates
# Run time: several minutes
generate_background_coordinates.py /path/to/bed_file.bed genome /path/to/output/
# Calculate motif scores for each sequence.
# Motifs used are specified individually.
# Most of the time, you'll probably want to use all of the default motifs, which can be specified with a wild card "*"
# The default motifs is a curated set of motifs formed from the JASPAR and CISBP motif databases.
# The default motifs are located at /path/to/tba/default_motifs
# Run time: ~15-20 minutes
create_features.py /path/to/output/fasta_file.fasta /path/to/output/background.fasta /path/to/output/ /path/to/tba/default_motifs/*
# Train the TBA model. This step will produce the weights/rankings for each motif as well as performance metrics for the model.
# Run time: ~2 minutes per cross-validation iteration
train_classifier.py /path/to/output/combined_features.tsv /path/to/output/labels.txt /path/to/output/
# Uses the likelihood ratio test to assign a significance level (p-value) to each motif
Run time: ~45 minutes per cross-validation iteration
calc_feature_significance.py /path/to/output/combined_features.tsv /path/to/output/labels.txt /path/to/output/
# Annotate model output with gene names
Run time: seconds
annotate_results_with_genes.py /path/to/output/coefficients.tsv /path/to/output/annotated_coefficients.tsv
annotate_results_with_genes.py /path/to/output/significance.tsv /path/to/output/annotated_significance.tsv
The final model outputs will be located at /path/to/output/. You should see several files:
/path/to/output/
background.bed
background.fasta
background_motif_scores.tsv
background_motif_starts.tsv
motif_scores.tsv
motif_starts.tsv
performance.tsv
coefficients.tsv
significance.tsv
Output file definitions are as follows:
- background.bed - genomic coordinates of background loci
- background.fasta - genomic sequence of background loci
- background_motif_scores.tsv - scores for each motif (columns) at each background loci
- background_motif_starts.tsv - starting position for each motif (columns) at each background loci
- motif_scores.tsv - scores for each motif (columns) at each loci of interest
- motif_starts.tsv - starting position for each motif (columns) at each loci of interest
- performance.tsv - performance of the model as measured by Precision (Column 1) and the area under the Receiver Operating Characteristic curve (Column 2) for each round of cross validation (rows).
- coefficients.tsv - the weight/coefficient assigned to each motif (rows) for each round of cross validation (columns)
- significance.tsv - the p-value assigned to each motif (rows) for each round of cross validation (columns)
To identify motifs of interest, you just need the coefficients.tsv and significance.tsv files. The remaining files may be useful for more in-depth customized analysis of your own design. The value of each weight indicates whether the presence of a motif is positively correlated (postive weights) or negatively correlated (negative weights) with your regions of interest. More important motifs tend to have weights with a larger magnitude. For example, if you trained a TBA model on the binding sites of the transcription factor CEBPa, the CEBPa motif should have a large positive weight. You can use the significance.tsv file to determine significant motifs. Start with a more stringent p-value threshold before considering more moderately ranked motifs; a p-value threshold of 10e-5 should be appropriate in most cases. Use of the mean value computed for each round of cross validation is recommended (average across the columns).
The outputs of the TBA model (significance, and coefficients) are only reliable if TBA can accurately discriminate regions of interest from background regions. You can assess the quality of a trained TBA model by looking at the performance.tsv file. TBA models that have a mean aucROC of at ~0.85 can be considered to be reasonably reliable. A model that randomly guesses would have an aucROC of 0.50.
Content coming soon - please refer to our BioRxiv manuscript for now for visualization ideas.
Content coming soon - please refer to our BioRxiv manuscript for now.
Content coming soon - please refer to our BioRxiv manuscript for now.
If you're ever unsure how to use a TBA command simply run the command without any parameters and help text should be displayed. All TBA commands and their associated parameters are listed here. Optional parameters are indicated with the default parameters
annotate_results_with_genes.py <result_path> <output_path>
Given a TBA coefficients file or a TBA significance file, maps the motif names to gene names
arguments:
* result_path - path to a TBA coefficients or significance file
* output_path - file path where output should be written
calc_feature_significance.py <feature_path> <label_path> <output_path> -num_iterations 5 -test_fraction 0.2 -num_procs 4
Performs an in silico mutagenesis test to assign significance to each motif
arguments:
* feature_path - path to a standardized feature set created by create_features.py
* label_path - path to a fasta_file containing negative sequences to score
* output_path - directory where output file should be written
optional arguments:
* -num_iterations - number of iterations to train classifier
* -test_fraction - fraction of data to use for testing classifier
* -num_procs - number of processors to use
calculate_all_motif_scores.py <fasta_path> <output_path> <motif_file_1> <motif_file_2>... -num_procs 4 -psuedocount 0.01
given a fasta file and a list and a list of motif files, calculates all matches to each motif for each sequence
arguments:
* fasta_path - path to a fasta_file containing sequences to score
* output_path - directory where output file should be written
* motif_files - list of motif files
optional arguments:
* -num_procs - number of processor cores to use
* -pseudocount - pseudocount for calculating motif scores
usage: create_features.py <positive_sequences_path> <negative_sequences_path> <output_path> <motif_file_1> <motif_file_2>... -num_procs 4 -pseudocount 0.01
Given a set of negative sequences and positive sequences (in FASTA format) as well as a list of motifs, calculates motif scores and sequence labels suitable for training classifier
arguments:
* positive_sequences_path - path to a fasta_file containing positive sequences to score
* negative_sequences_path - path to a fasta_file containing negative sequences to score
* output_path - directory where output file should be written
* motif_files - list of motif files
optional arguments:
* -num_procs - number of processor cores to use
* -pseudocount - pseudocount for calculating motif scores
extract_sequences.py <bed_file> <output_file_path>
Extracts the genomic sequence at positions indicated by input bed file
arguments:
* bed_file - path to a bed file
* genome - build of genome to use (eg. hg38, mm10)
* output_file_path - directory where output file should be written
generate_background_coordinates.py -sizeRatio 1 -numBins 10 -nThreshold 0.1 -filterChromosomes chrM chrY
Constructs random GC matched background regions
arguments:
* inputPath - path to a bed file containing a chr, start, end, and strand column
* genome - genome from which to construct background regions
* outputPath - directory where output files should be written
optional arguments:
* -sizeRatio - size of the background region with respect to the target region
* -numBins - number of bins to use for GC normalization
* -nThreshold - maximum fraction of background sequences that can be N
* -filterChromosomes - chromosomes to ignore
train_classifier.py <feature_path> <label_path> <output_path> -num_iterations 5-test_fraction 0.2
Given standardized motif features and matching labels trains a classifier and returns performance metrics and model coefficients
arguments:
* feature_path - path to a standardized feature set created by create_features.py
* label_path - path to a fasta_file containing negative sequences to score
* output_path - directory where output file should be written
optional arguments:
* -num_iterations - number of iterations to train classifier
* -test_fraction - fraction of data to use for testing classifier
train_model_default.sh <bed_file>
Wrapper script for training a TBA model to distinguishes sequences in input bed file from genomic background with default parameters
arguments:
* bed_file - input bed file
* genome - genome build
* output_directory - directory where file outputs should be written
optional arguments:
* -t - generate scripts but do not execute them
train_comparison_model.sh <bed_file_1> <bed_file_2>
Wrapper script for training a TBA model to distinguishes sequences in bed_file_1 from bed_file_2 from genomic background with default parameters
arguments:
* bed_file_1 - input bed file
* bed_file_2 - input bed file
* genome - genome build
* output_directory - directory where file outputs should be written
optional arguments:
* -t - generate scripts but do not execute them
TBA was created by Jenhan Tao with feedback from Gregory Fonseca and Christopher Benner. If you have any questions, please send an email to jenhantao@gmail.com. We would be glad to help you apply TBA to your research problem
If you use TBA for research, please cite the following manuscript:
Fonseca, G. J., Tao, J. Diverse motif ensembles specify non-redundant DNA binding activities of AP-1 family members in macrophages. Preprint at: https://www.biorxiv.org/content/early/2018/06/13/345835 (2018).
MIT License
Copyright (c) 2017 Jenhan Tao
Permission is hereby granted, free of charge, to any person obtaining a copy
of this software and associated documentation files (the "Software"), to deal
in the Software without restriction, including without limitation the rights
to use, copy, modify, merge, publish, distribute, sublicense, and/or sell
copies of the Software, and to permit persons to whom the Software is
furnished to do so, subject to the following conditions:
The above copyright notice and this permission notice shall be included in all
copies or substantial portions of the Software.
THE SOFTWARE IS PROVIDED "AS IS", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR
IMPLIED, INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY,
FITNESS FOR A PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE
AUTHORS OR COPYRIGHT HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER
LIABILITY, WHETHER IN AN ACTION OF CONTRACT, TORT OR OTHERWISE, ARISING FROM,
OUT OF OR IN CONNECTION WITH THE SOFTWARE OR THE USE OR OTHER DEALINGS IN THE
SOFTWARE.