Overall Workflow
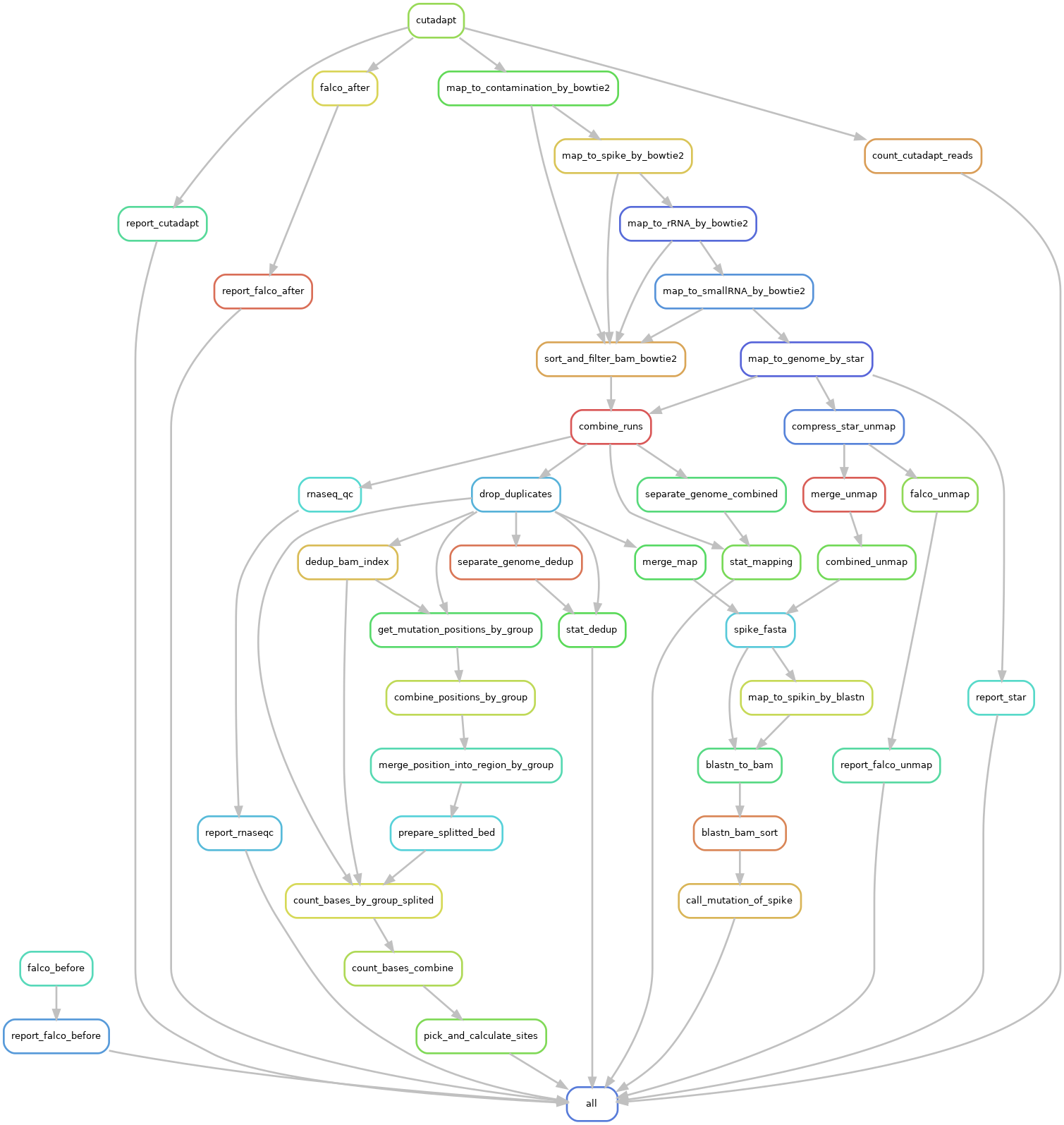
We will first perform read trimming to remove adapter, primer sequences, molecular barcode and low quality bases using cutadapt software. A more stringent quality filtering that uses the fastp software will also be applied to filter out low quality sequences and generate the quality report.
After Quality control, each trimmed read contains a high quality insert fragment and a unique molecular index (UMI). All trimmed reads will be mapped to E. coli and Mycoplasma genome to filter biological contamination RNA using the bowtie2 Tool, and then unmapped reads will be mapped to spike-in sequence to filter RNA spike-in. Similarly, unmapped reads will be mapped to the ribosomal RNA, small RNA and mRNA (whole genome) reference sequence sequentially. After mapping, reads with identical UMI and mapped to the same location will be treated as PCR duplicates and dropped from downstream analysis.
We detect mutation site by site on multiple samples simultaneously, and calculates mutation number and sequencing depth for each mutation position among all the samples. Then putative m6A sites will be detected based on the mutation ratio, mutation number and sequencing depth of each mutation position.